Michelle S. Yau1, Paul C. Okoro2, Ida K. Haugen3, John A. Lynch4, Michael Nevitt5, Cora E. Lewis6, James Torner7 and David Felson8, 1Hebrew SeniorLife, Harvard Medical School, Boston, MA, 2Hebrew SeniorLife, Boston, MA, 3Center for treatment of Rheumatic and Musculoskeletal Diseases (REMEDY), Diakonhjemmet Hospital, Oslo, Norway, Oslo, Norway, 4UCSF, San Francisco, CA, 5University of California at San Francisco, Orinda, CA, 6University of Alabama at Birmingham, Birmingham, AL, 7University of Iowa, Iowa City, IA, 8Boston University, Boston, MA
Background/Purpose: Epigenetic mechanisms like DNA methylation may play a role in OA. For example, expression of GDF5, a well-known OA gene, is modulated by demethylation of the 5' untranslated region. There may be other loci under epigenetic regulation that could provide mechanistic insights into OA. To our knowledge, there are no epigenome-wide association studies of OA that have been carried out in blood or in large cohorts. We therefore aimed to identify differentially methylated loci in blood that are associated with OA.
Methods: We analyzed data from the Multicenter Osteoarthritis Study (MOST), a longitudinal, prospective study of knee OA. A subset of participants ranging in age from 45-69 years were recruited in 2016 with Kellgren-Lawrence grade (KL) 0, 1, or 2 in the worse affected of the tibiofemoral or patellofemoral compartments in both knees and no continuous and severe pain in either knee. We assessed follow-up data on these participants collected in 2018, where we assessed DNA methylation in 671 participants who had blood samples and weight-bearing fixed flexion posteroanterior (PA) and lateral knee radiographs and bilateral PA hand radiographs. Knee and hand joints were read for KL grade by a team of readers with adjudication. We measured DNA methylation with the Illumina Infinium MethylationEPIC 850K array. We removed poor quality probes and samples, then conducted stratified quantile normalization. We additionally removed probes overlapping single nucleotide polymorphisms, probes that were cross-reactive and probes on sex chromosomes. This resulted in 671 samples and 774,460 CpGs for analysis. CpG methylation beta-values were log2 transformed to M-values for statistical analyses.
We assessed associations between each CpG and hand, knee, and multi-joint OA using linear mixed effects regression. Models were adjusted for age, sex, smoking status, race, and estimated blood cell counts as fixed effects and array identifiers including sample plate number and row number as random effects. Hand OA was defined as the presence of 1) three or more hand joints with KL grade ≥ 2 where at least two are part of the same DIP, PIP, or CMC1 joint group across both hands, 2) KL grade ≥ 2 in at least one DIP, and 3) bilateral involvement in ≥ 1 of the DIP, PIP, or CMC1 joint groups. Knee OA was defined as KL grade ≥ 2 in at least one knee. Multi-joint OA was defined as the presence of both hand and knee OA.
We set the p-value threshold for epigenome-wide significance to 1 × 10-6 as suggested by Tsai and Bell (2015). All analyses were conducted in R.
Results: Mean age was 58±7 years and 53% were women. About 23% had hand OA, 25% had knee OA, and 8% had multi-joint OA. We identified one CpG probe associated with knee OA and four CpG probes associated with multi-joint OA; none were associated with hand OA (Table 1). Associated CpG probes were specific to each OA phenotype assessed.
Conclusion: We identified five differentially methylated loci associated with OA, the majority of which were associated with multi-joint OA, underscoring its possible systemic etiology. Our top finding, PTPRN2, is a gene that encodes a protein tyrosine phosphatase receptor that may be involved in the regulation of insulin secretion. Replication of these findings in independent cohorts is ongoing. Table 1. CpGs significantly associated with OA (P-values < 1E-6) Disclosures: M. Yau, None; P. Okoro, None; I. Haugen, None; J. Lynch, None; M. Nevitt, None; C. Lewis, None; J. Torner, None; D. Felson, None.


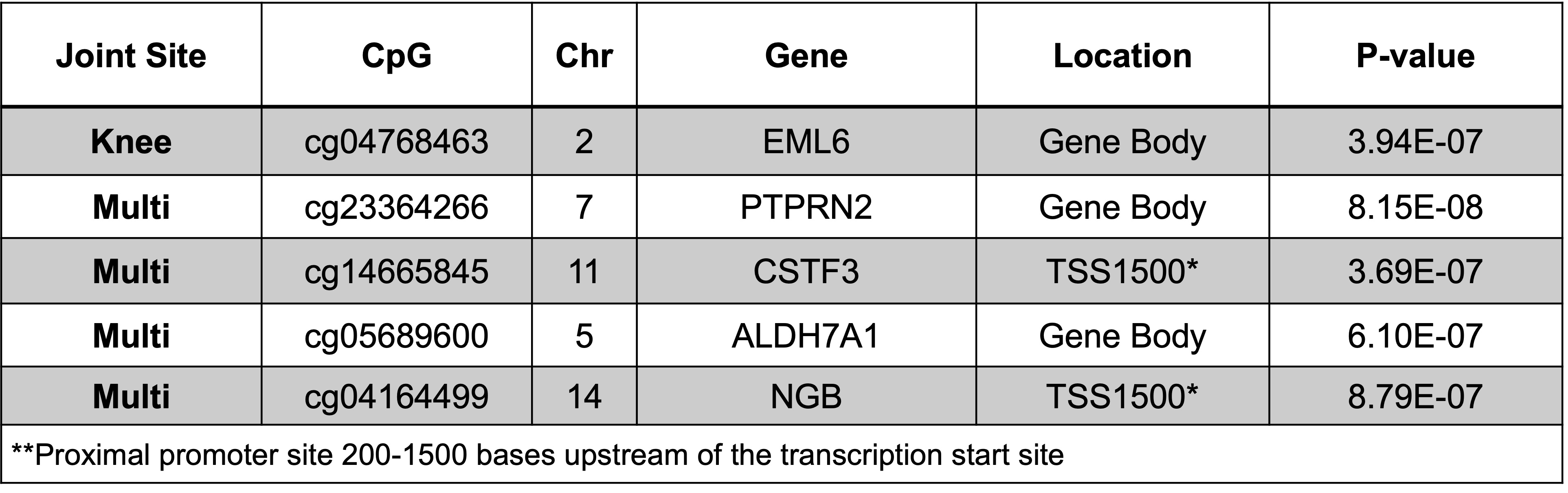 Table 1. CpGs significantly associated with OA (P-values < 1E-6)
Table 1. CpGs significantly associated with OA (P-values < 1E-6)