
Poster Session D

Monique Hinchcliff, MD, MS
Yale School of Medicine
Westport, CT, United States
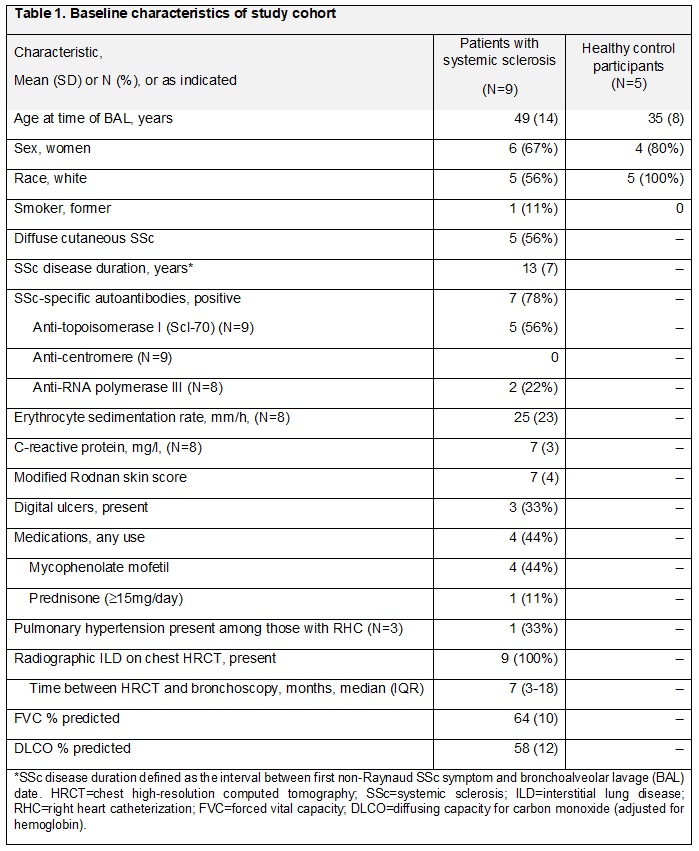
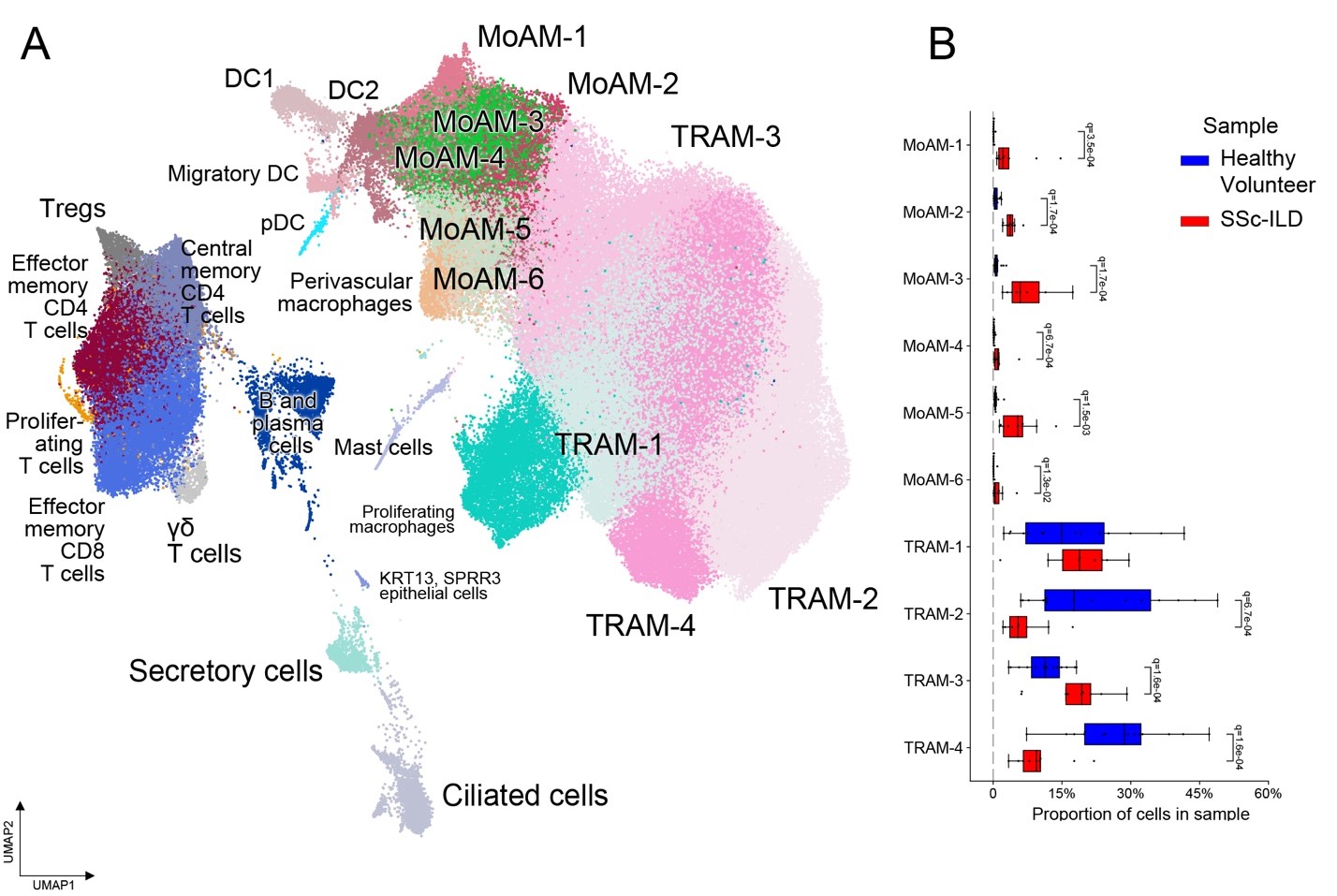 Figure 1: Monocyte-derived alveolar macrophages are expanded in SSc-ILD patients. (A) UMAP representation of cell transcriptional profiles from 25 BAL samples from 9 SSc-ILD patients, 5 healthy volunteers and 10 external published healthy volunteers, colored by cell subtype; (B) proportion of cells from TRAM and MoAM subtypes in samples.
Figure 1: Monocyte-derived alveolar macrophages are expanded in SSc-ILD patients. (A) UMAP representation of cell transcriptional profiles from 25 BAL samples from 9 SSc-ILD patients, 5 healthy volunteers and 10 external published healthy volunteers, colored by cell subtype; (B) proportion of cells from TRAM and MoAM subtypes in samples.