
Ignite Talk
Katherine Owen, PhD
RILITE
Charlottesville, VA, United States
Disclosure: Disclosure information not submitted.
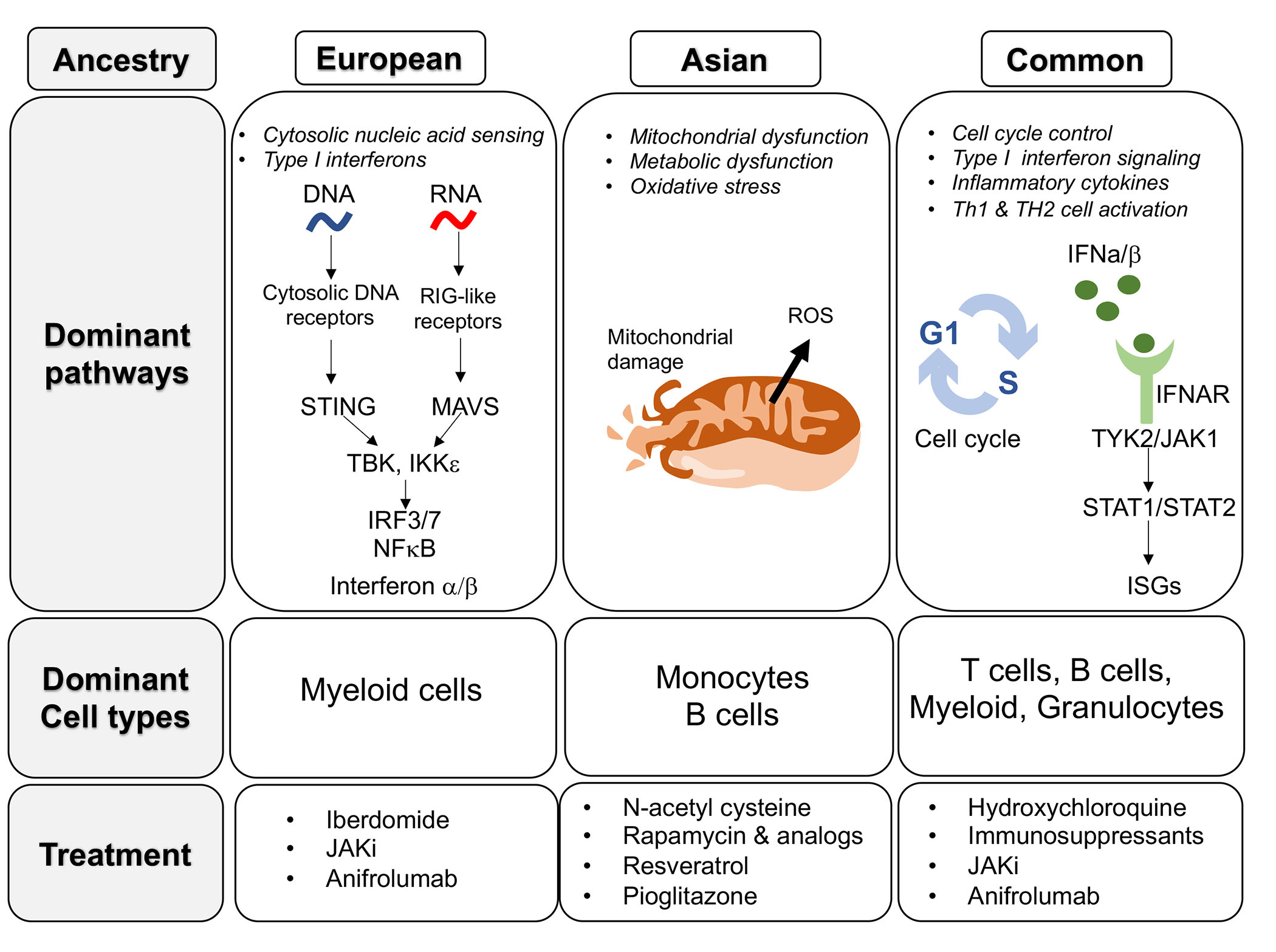 Figure 1. Summary of lupus molecular pathways, dominant cell types and potential treatment options in EA and AsA ancestral populations.
Figure 1. Summary of lupus molecular pathways, dominant cell types and potential treatment options in EA and AsA ancestral populations.