Purdue University West Lafayette, Indiana, United States
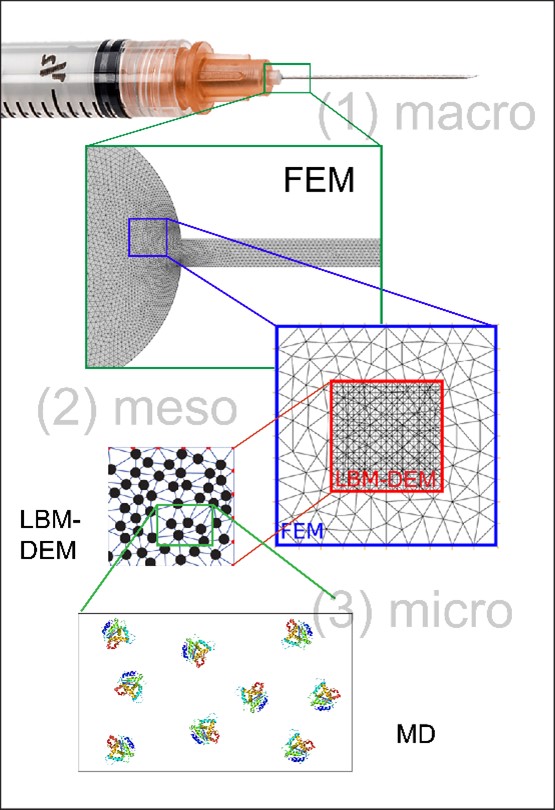
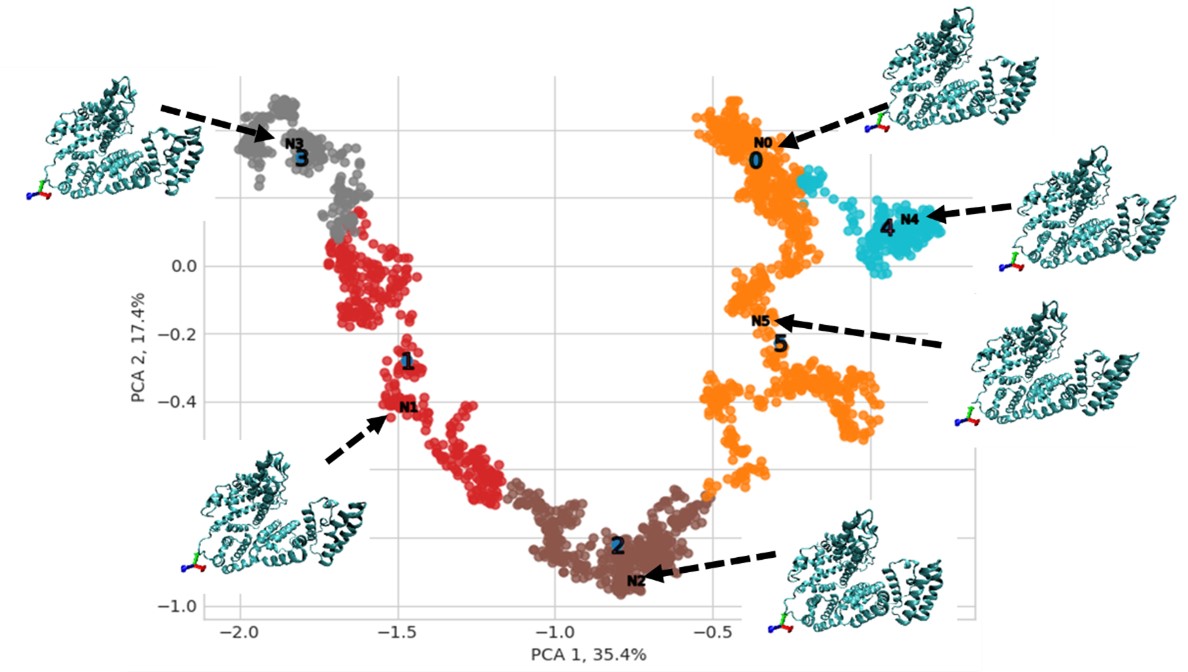
Purpose: The high specificity of proteins in biological processes, makes them perfect candidates for pharmacotherapeutics. As a result, the use of biotherapeutics is on the incline in several areas of therapy that are traditionally for small molecules. However, this growth is hampered by stability issues that occur due to complex protein-protein interactions. Many of the industry-based techniques for protein stability studies are time consuming and insufficient for capturing the complexities of protein interactions. Computational modeling, more specifically, molecular dynamics simulations, are purported to have the advantage of accessing and sampling more thermodynamic states (than humanly achievable) of proteins in various environments; sampling of those states provides information about the free unfolding energy of proteins under mechanical and hydrodynamic stress. Consequently, a multiscale modeling of proteins under hydrodynamic stress with molecular dynamics, will inform the design space (formulation, manufacturing, and administration) of protein therapeutics by providing the potential of mean force associated with the conformational space of proteins under stress. Methods: Three ‘scales’ of simulations are used to model protein-protein interactions. Figure 1 shows a complete schematic view of the multiscale simulation model. The first is a macroscale simulation where solvated proteins are treated implicitly as a fluid continuum to assess the effects of fluid velocity and shear stress using Finite Element Method (FEM). The velocity effects are then used in a mesoscale simulation where proteins are treated as individual particles by coupling the Lattice Boltzmann Method (LBM) with Discrete Element Method (DEM). This step simulates the behavior of protein particles in different environments and provides the effect of particle collisions on the kinetic energy of each particle. Finally, kinetic perturbations (via thermal ramping) of the particles are carried out using molecular dynamics (MD) in a microscale simulation. This provides the trajectory of the protein conformations that are then clustered and averaged. The average conformation(s) from the clusters can be assessed with adaptive biasing forces (ABF) to determine the free energy surface of the protein conformation(s) along a distance-based reaction coordinate. Results: Cluster analysis of the simulation trajectory obtained from MD simulations provided the average conformations of the proteins under hydrodynamic stress. The average protein structures revealed unfolding along the reaction coordinates. Figure 2 shows the average protein structures along a reaction trajectory for a model protein. Conclusion: Multiscale modeling of protein-protein interactions provides a complete analysis of the conformational space of proteins under hydrodynamic stress. The averaged conformation(s) can be assessed for the energy minima and free unfolding energy, and provide valuable information about formulation and device design strategies that can avoid unfavorable protein interactions. References: Lei Xing, Yue Li, and Tonglei Li, PDA Journal 2019, 73, 260-275
Acknowledgements: This project is funded by the Center for Pharmaceutical Processing Research (CPPR)
Figure 1: Multiscale simulation modeling: The macroscale simulation uses FEM and treats the protein solution as a continuous fluid phase (Lei Xing, Yue Li, and Tonglei Li, PDA Journal 2019, 73, 260-275); the mesoscale simulation uses the velocity effects of the fluid flow to find the effects of particle collision on the kinetic energy of the particles using LBM and DEM; the microscale simulation use MD to find the average conformation(s) found in the trajectory of kinetically perturbed particles.
Figure 2: Simulation trajectory and average structures found after cluster analysis of a model protein (BSA): PCA is used for dimensionality reduction to map the trajectory of the simulation. Cluster analysis is used to find the average conformations along the trajectory. For the model protein, there are six average conformations.