Purdue University West Lafayette, Indiana, United States
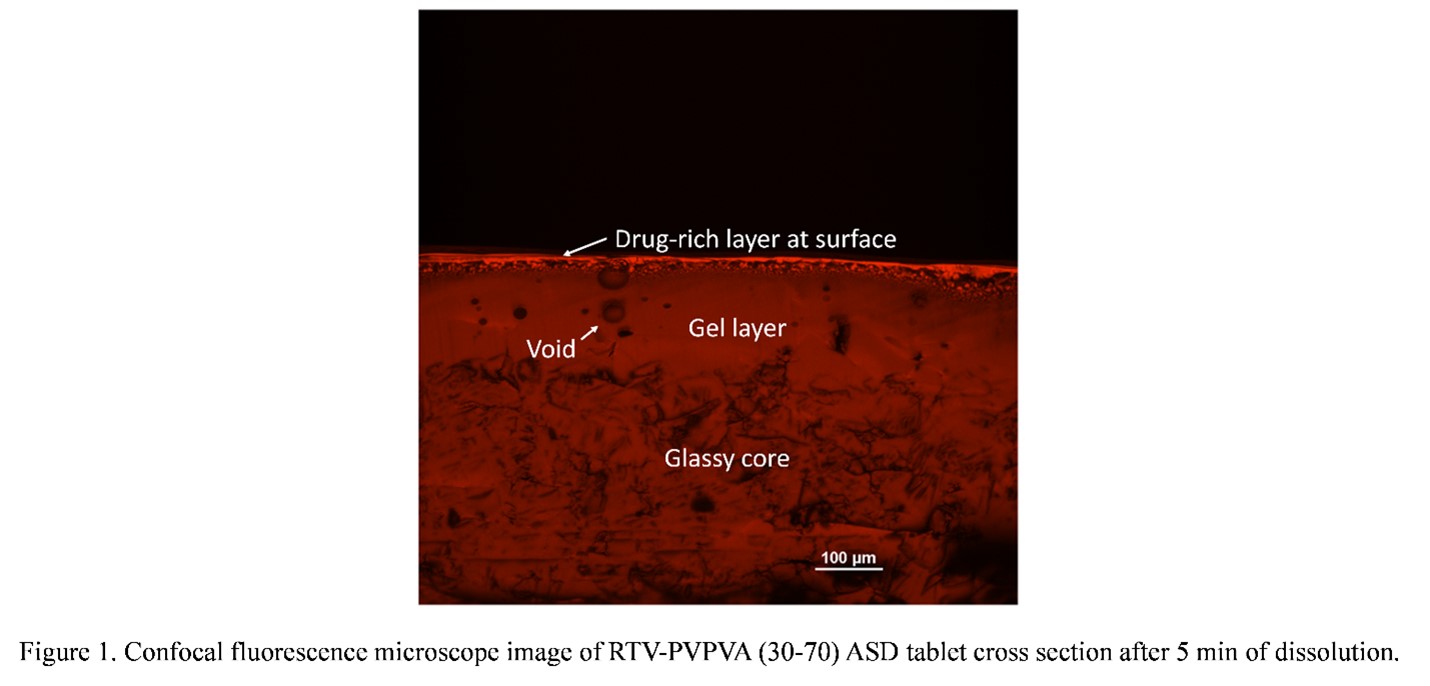
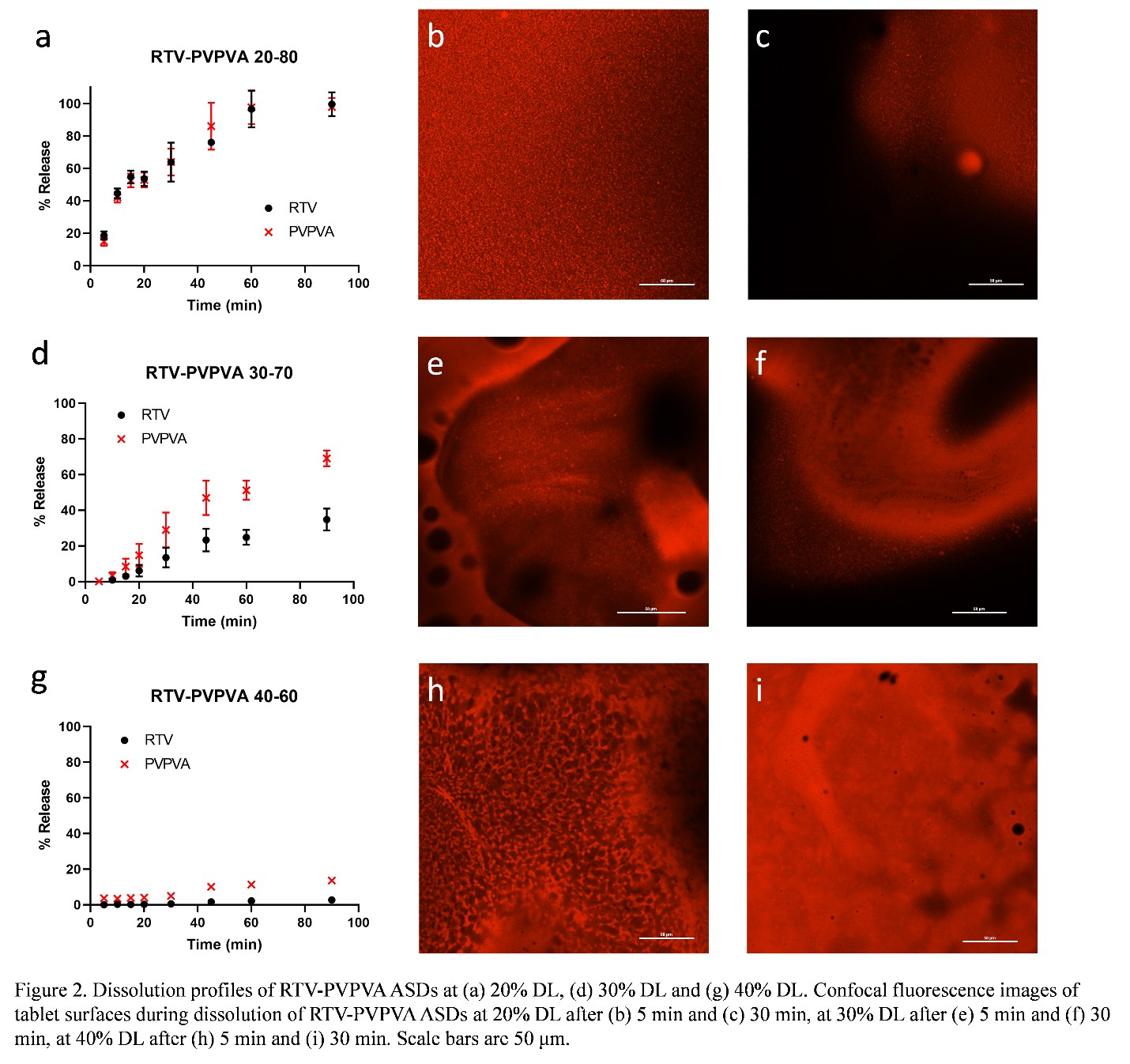
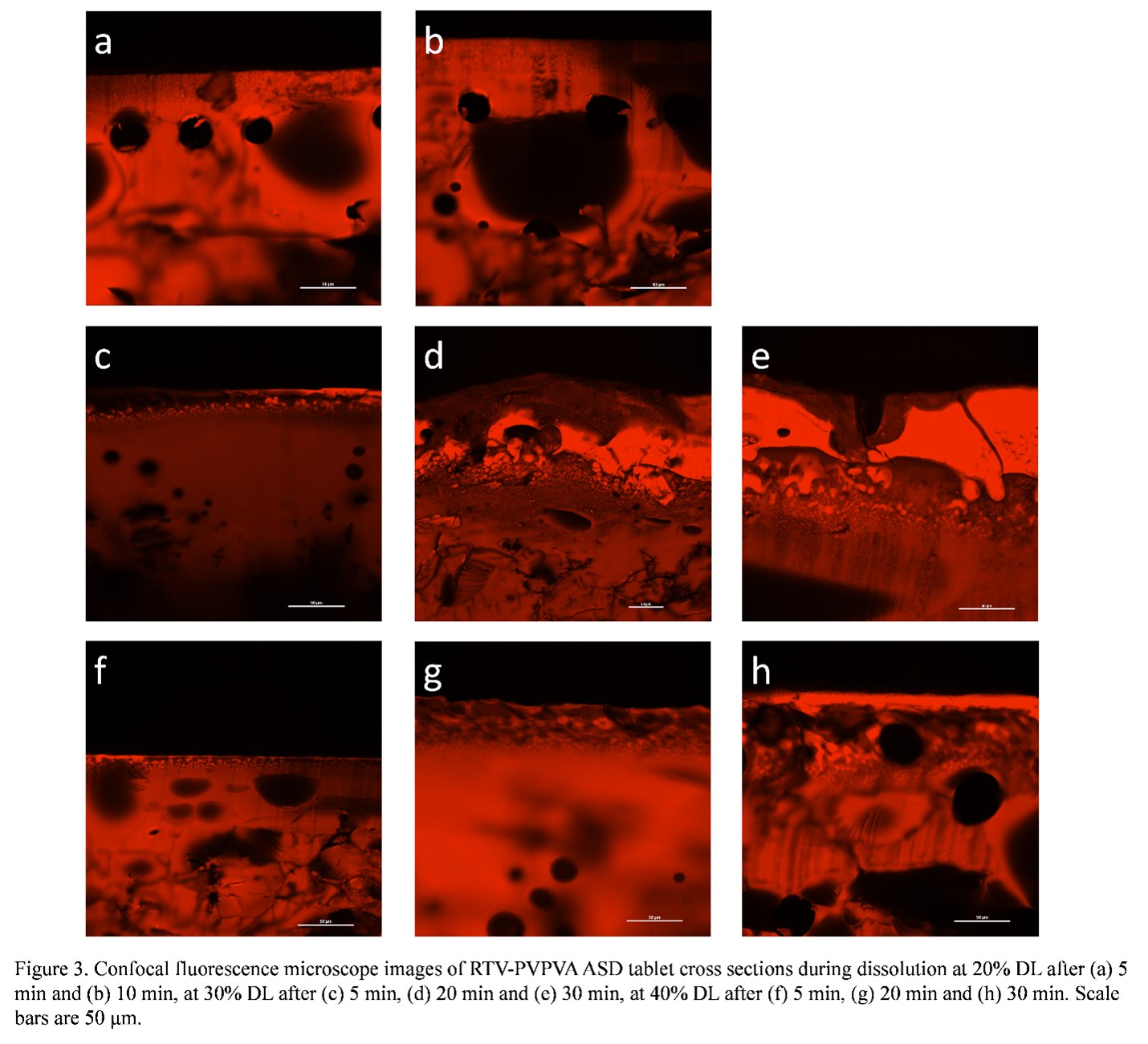
Purpose: With increasing numbers of new chemical entities (NCEs) with poor aqueous solubility entering the product pipeline, formulation strategies are often needed to address their intrinsically low solubilities which can lead to poor oral absorption and bioavailability.1 Amorphous solid dispersion (ASD), where an amorphous drug and polymer are molecularly mixed,2 has shown great potential to address these concerns. Dissolution of ASDs can result in rapid dissolution rates as well as apparent drug concentrations that exceed the drug amorphous solubility. However, desirable dissolution performance is only achieved at relatively low drug loadings (DLs) and drug release tends to decrease with increasing DL. Poor release at higher DLs has been associated with the occurrence of amorphous-amorphous phase separation (AAPS) during ASD dissolution,3 a process where an initially miscible ASD system separates into drug-rich and polymer-rich phases upon hydration. In contrast, others have proposed that discrete drug-rich domains are created by AAPS pre-dissolution.4 Therefore, the impacts of AAPS on ASD dissolution are rather complicated and both the kinetics and phase morphology should be considered. Thus, it is important to investigate the effects of AAPS on ASD dissolution to better understand their dissolution mechanism. The objective of this study is to understand different AAPS behavior during dissolution at different DLs and corelate their solid-state phase behavior to the dissolution behavior. Methods: Ritonavir (RTV) and polyvinylpyrrolidone/vinyl acetate (PVPVA) ASDs at different drug loadings (DLs) were prepared with rotary evaporation. ASD dissolution behavior was studied with surface normalized dissolution. Phase separation morphology on partially dissolved ASD compact surfaces was investigated using a confocal fluorescence microscope, with Nile red as the fluorescence marker. Tablets were also cut in half with a razor blade to observe the compact cross sections using confocal microscopy. Results: During ASD compact dissolution, a gel layer is formed at the interface between the glassy compact (which shows individual particles and grain boundaries) and the dissolution media as shown in Figure 1. This is a homogeneous layer formed by plasticization of ASD matrix by water to above the glass transition temperature. Voids are present in this layer due to entrapment of air during compression to form the compact. AAPS was observed near the gel-solution interface for all DLs studied. However, the phase separation behavior varied significantly at different DLs. At a low DL (20%), complete and congruent release of drug and polymer was observed (Figure 2a). At this DL, discrete drug-rich domains were formed at dissolving compact surfaces throughout the dissolution process, as shown in the confocal fluorescence images taken on the tablet surfaces after dissolution times of 5 and 30 min (Figure 2b, c). The discrete drug-rich domains were also observed in the cross-section images for at the same DL (Figure 3a, b) during dissolution. At a high DL (40%), no release of drug or polymer was observed (Figure 2g). A continuous drug-rich domain that evolved into a drug-rich layer covering the dissolving compact surfaces at later time points was observed at this DL (Figure 2h, i). On the cross-section images, the same morphology was observed where the compact surface was covered with a drug-rich layer that increases in thickness with time (Figure 3f-h). This drug-rich layer on the compact surface prevented any release of drug or polymer. For an intermediate DL (30%), both discrete and continuous drug-rich domains were observed on compact surfaces (Figure 2e, f) or cross sections (Figure 3d, e), which explained the partial drug release (Figure 2d). Conclusion: The formation of a continuous drug-rich phase after AAPS at the compact-solution interface is detrimental to drug release from ASD systems. Such a phase behavior is responsible for the poor drug release at high DLs for the RTV-PVPVA ASD system studied. References: 1. Williams, H. D.; Trevaskis, N. L.; Charman, S. A.; Shanker, R. M.; Charman, W. N.; Pouton, C. W.; Porter, C. J. H., Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65 (1), 315-499. 2. Huang, Y.; Dai, W. G., Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B. 2014, 4 (1), 18-25. 3. Indulkar, A. S.; Lou, X.; Zhang, G. G. Z.; Taylor, L. S., Insights into the dissolution mechanism of ritonavir-copovidone amorphous solid dispersions: Importance of congruent release for enhanced performance. Mol. Pharm. 2019, 16 (3), 1327-1339. 4. Li, N.; Taylor, L. S., Microstructure formation for improved dissolution performance of lopinavir amorphous solid dispersions. Mol. Pharm. 2019, 16 (4), 1751-1765.
Acknowledgments: Ruochen Yang would like acknowledge ASTAR, Singapore, for funding. HSP, and GGZZ, are employees of AbbVie and may own AbbVie stock. AbbVie sponsored and funded the study; contributed to the design; participated in the collection, analysis, and interpretation of data, and in writing, reviewing, and approval of the final publication.