Purpose: Inhibitors of Bruton’s Tyrosine Kinase (BTK) and proteasome inhibitors are currently used to treat hematologic malignancies. While the ubiquitin-proteasome system is necessary for protein homeostasis in all mammalian cells, BTK is unique to some B-cell malignancies. BTK inhibitor ibrutinib was previously shown to synergize with proteasome inhibitors in myeloma and mantle cell lymphoma cells. that inhibit proteasome β5 sites and with LU-102, a specific inhibitor of the β2 site of the proteasome; the synergy with β2-specific LU-102 was stronger than with the FDA-approved β5 inhibitors carfilzomib and bortezomib. However, the synergy was not dependent on BTK expression and occurred at >100-fold higher concentration than is needed for complete inhibition of BTK. In order to identify the mechanism of this off-target effect, we explored combinations of LU-102 with Ibrutinib and other BTK inhibitors in triple-negative breast cell (TNBC) lines that do not express BTK but are sensitive to proteasome inhibitors. TNBC confers poor prognosis and lacks targeted therapies. We found that two allosteric BTK inhibitors, GDC-0853 and CGI-1746, were more synergistic with LU-102 than ibrutinib and other competitive BTK inhibitors. Surprisingly CGI-1746, while not toxic to these cell lines as a single agent, induced robust apoptosis when combined with LU-102. Our aim was to identify the target(s) of BTK inhibitors responsible for synergy with LU-102 in TNBC. Methods: After our preliminary viability assays and caspase activation assays that confirmed synergy, we used RNA sequencing to identify pathways changed upon treatment with BTK inhibitors in TNBC. After changes in pathways pertaining to protein ubiquitination and unfolded protein response were identified as being upregulated, we performed western blotting and enzyme activity assays that confirmed proteasome inhibition by BTK inhibitors. Results: We found by RNA sequencing that unfolded protein response and heat shock response were upregulated in response to the treatment with the CGI-1746 and LU-102 combination. And that combinations of CGI-1746 and ibrutinib with LU-102 and carfilzomib caused synergistic accumulation of ubiquitylated proteasome substrates. CGI-1764, GDC-8053, and Ibrutinib inhibited all the catalytic subunits of the proteasome 20S proteolytic core in a non-competitive, allosteric manner. Furthermore, CGI-1746, but not other BTK inhibitors, inhibited the ATPase activity of 26S proteasome at concentrations that cause synergy with LU-102. Thus, several BTK inhibitors inhibit the proteasome by a novel mechanism, and CGI-1746 is the first inhibitor of proteasomal ATPases. Conclusion: Our findings with CGI-1746 and other kinase inhibitors suggest a need for more stringent screening of kinase inhibitors for interaction with non-kinase targets such as the proteasome as off-targets. Our findings also show the importance of developing allosteric inhibitors of the proteasome catalytic sites and specific inhibitors of the proteasome ATPases to sensitize cancer cells to currently approved inhibitors of the proteasome β5 site. References: 1. Kraus, J. et al. The novel β2-selective proteasome inhibitor LU-102 decreases phosphorylation of I kappa B and induces highly synergistic cytotoxicity in combination with ibrutinib in multiple myeloma cells. Cancer Chemother. Pharmacol. 76, 383–396 (2015).
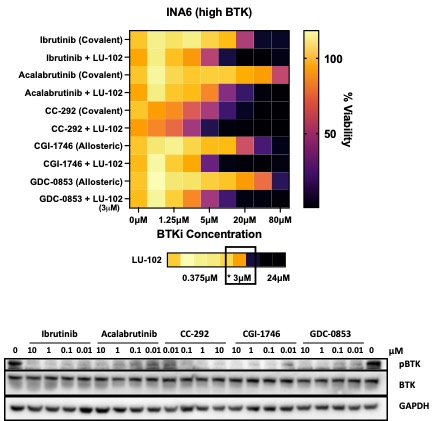
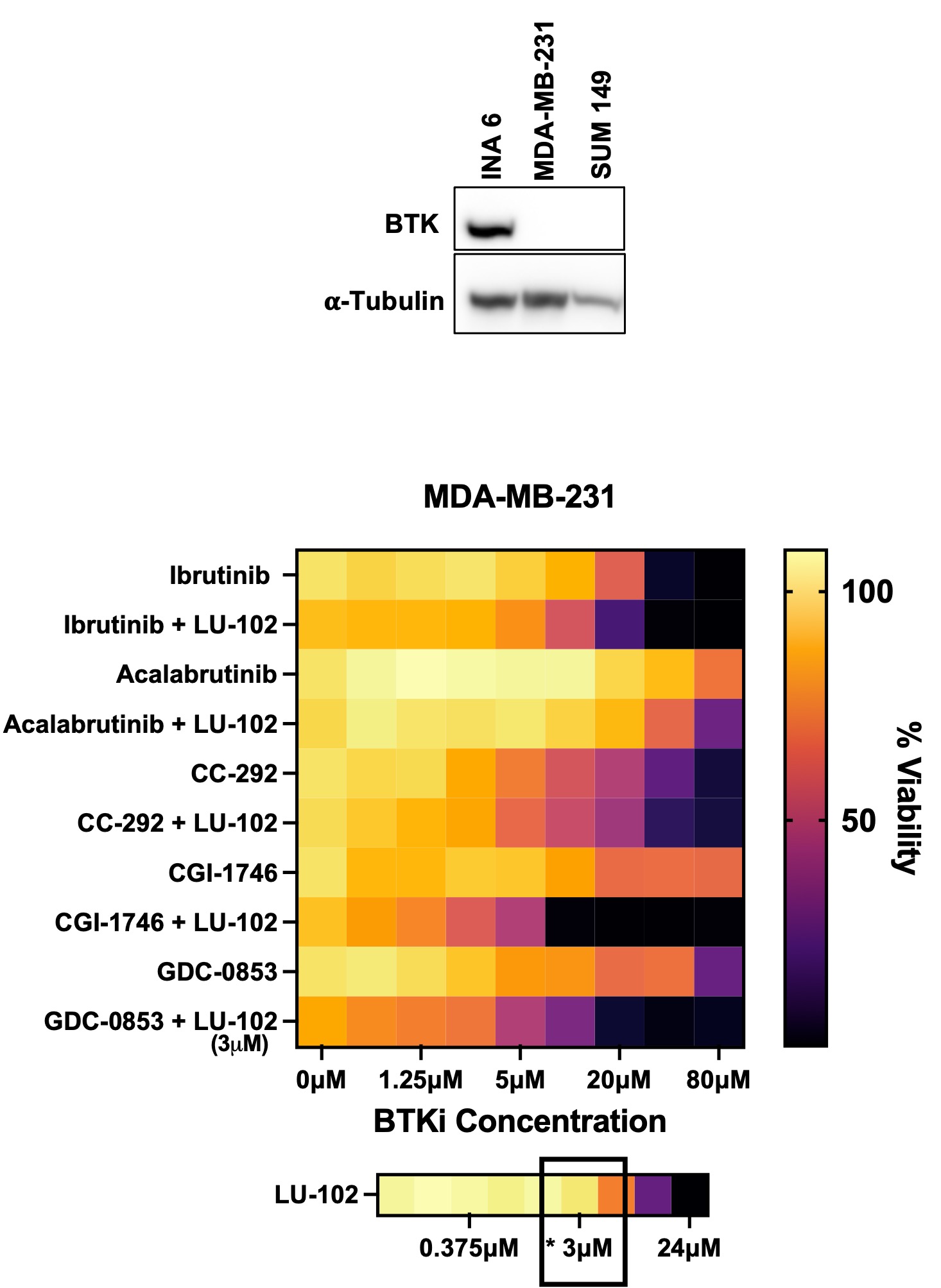
Synergy with LU-102 is not dependent on BTK inhibition by BTK inhibitors
Some BTK inhibitors synergyze with LU-102 in TNBC cells which do not express BTK
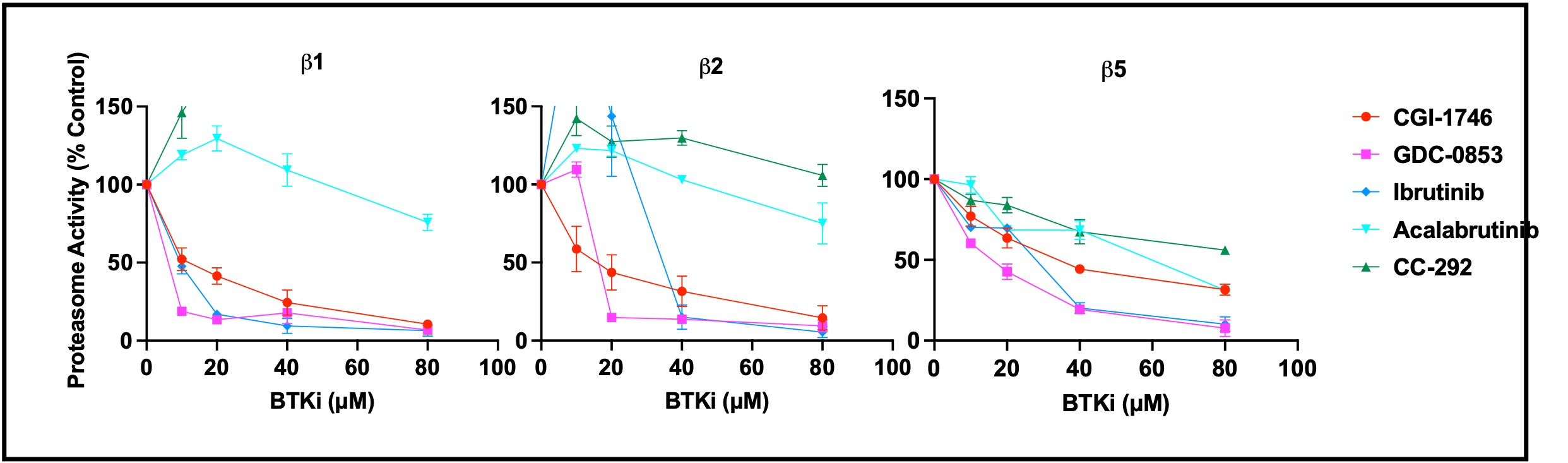
BTK inhibitors which synergize with LU-102 inhibit all three proteasome active sites