Postdoctoral Associate Duke University Medical Center
Introduction: Using a genetic mouse model of Type 1 diabetes that develops bladder underactivity, we have explored several molecular pathways that lead to emptying inefficiency. One such target lies within the prostaglandin (PG) signaling pathways. PGs are synthesized within urothelia and modulate bladder contractility by binding receptors on the detrusor. Although diabetes downregulates urothelial PG production, little is known about the responsiveness of the diabetic detrusor to PG stimulation. Here, we used ex vivo myography of denuded bladder strips to examine detrusor contractility in response to PGE2 and PGF2a, the main PGs produced by the urothelia and agonists of EP and FP receptors, respectively. We have further investigated a role for inflammation in any changes by examining urothelia from diabetic mice with NLRP3 genetically deleted.
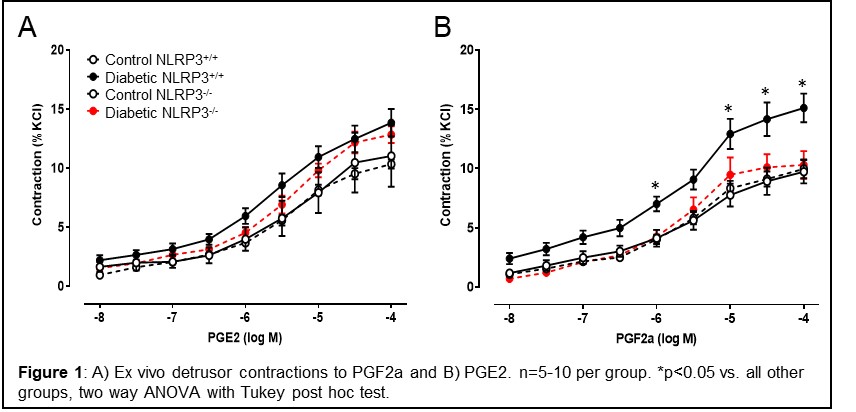
Methods: Type 1 diabetic Akita mice were crossbred with NLRP3-/- mice to yield 4 groups of mice: control/NLRP3+/+, diabetic/NLRP3+/+, control/NLRP3-/-, and diabetic/NLRP3-/-. Females were aged to 30 weeks when UAB is apparent. For ex vivo analysis, strips of denuded bladders were mounted in myograph chambers and maintained in aerated 37°C Krebs solution. Concentration response curves to PGE2 and PGF2a were constructed (n=5-10 per group).
Results: PGE2 induces dose-dependent contractions in control detrusors that are slightly, but not significantly, increased in the diabetic detrusors. Detrusors from diabetic/NLRP3-/- mice (i.e. diabetic mice but without inflamed bladders) were not significantly different. In contrast, contractions to PGF2a, a FP receptor agonist, are significantly higher in detrusors from diabetic/NLRP3+/+ mice (p < 0.05). These stronger contractions were completely absent in detrusors from diabetic mice lacking NLRP3 (i.e. diabetic mice but without inflamed bladders).
Conclusions: Detrusor contractility in response to PGF2a is increased in diabetic mice with UAB and this is driven by NLRP3-induced inflammation. This increased sensitivity may make PGF2a, or other FP receptor agonists, an effective therapy to treat existing UAB.
Source of Funding: NIH-K12 DK100024; NIH-RO1 DK117890

.jpg)
.jpg)