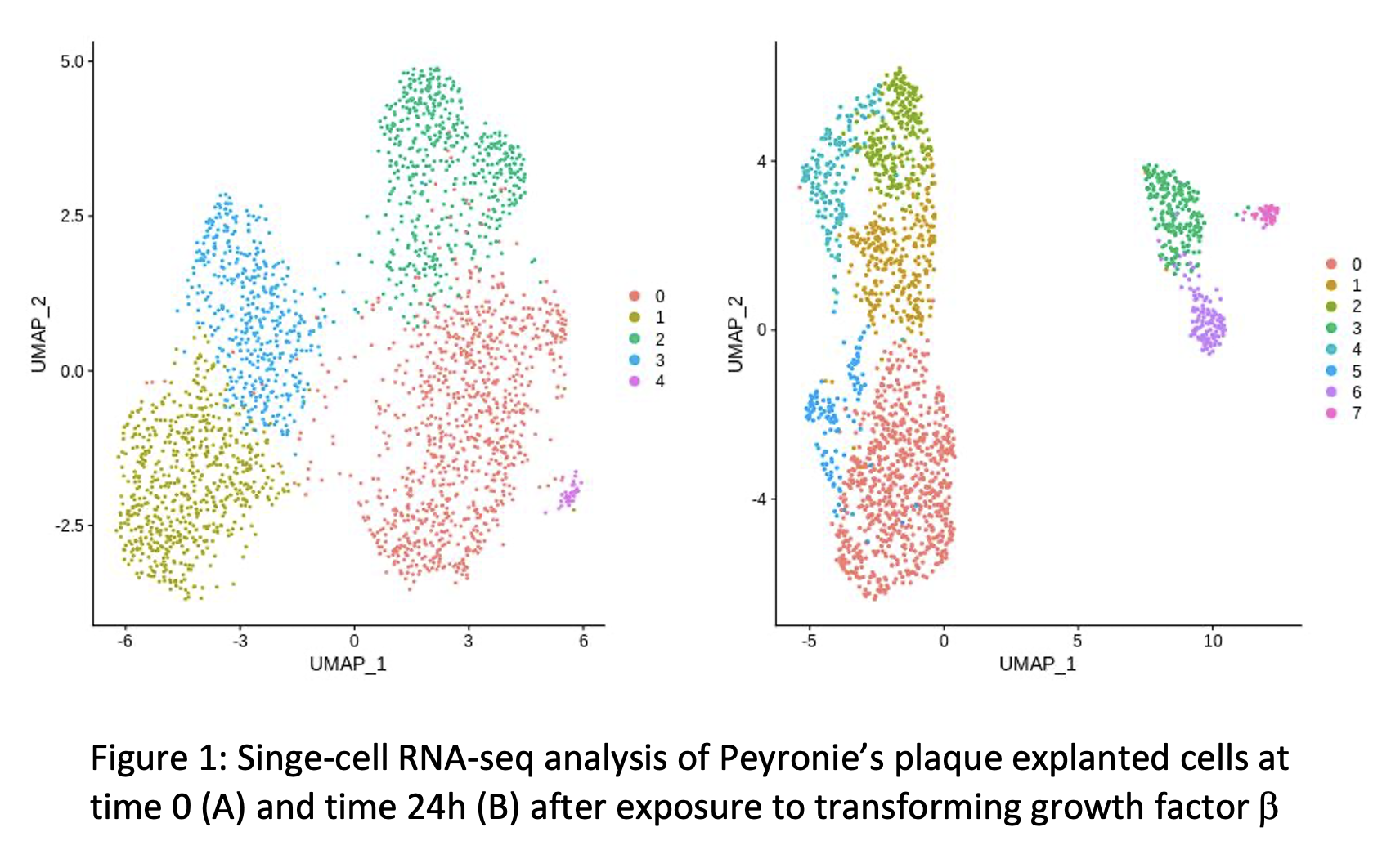
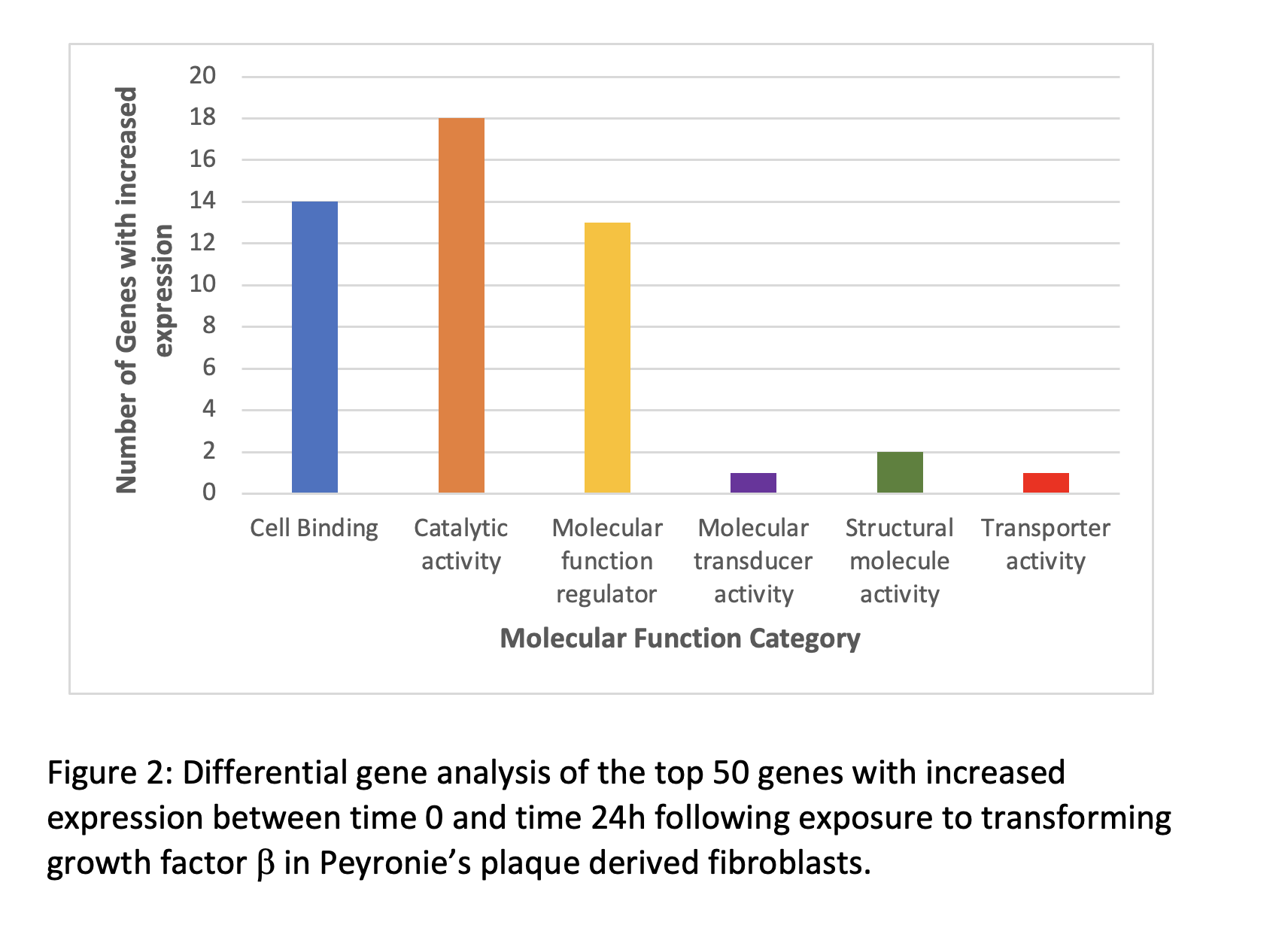
Introduction: The mechanisms behind the pathogenesis of Peyronie’s disease largely remain a mystery. Single-cell transcriptomics provides a new means to assess the genetic expression profiles occurring on a cell-by-cell basis within a Peyronie’s plaque.
This study set out to use single-cell RNA sequencing to investigate the transcriptional pathways involved in Peyronie’s disease.
Methods: Tunica albuginea biopsy was performed in a patient undergoing penile incision and grafting surgery. Cells were explanted from biopsy tissue and propagated. Peyronie’s plaque derived cells were then exposed to transforming growth factor beta(TGF-beta). RNA-seq single cell sequencing was then completed at 0 and 24 hours. We used principle component analysis (PCA) to reduce the dimension of data and built the nearest-neighbors graph using PCA weights. We performed clustering over the nearest-neighbor graphs and visualized the clusters using UMAP. Differential gene expression analyses were performed using the top 50 genes with increased expression between time points.
Results: Assessment of cell clustering identified several potential cell populations that became more clearly defined following administration of TGF-beta (Figure 1). Known gene expression profiles of fibroblasts, myofibroblasts, and smooth muscle were applied to our cluster analysis and did not localize to specific cell cluster populations suggesting myofibroblasts were largely present at both time points.
Differential gene expression showed that genes involved in cellular binding and catalytic activity were most commonly upregulated (Figure 2).
Conclusions: This analysis provides insight into the expression profiles of Peyronie’s plaque cell populations stimulated via TGF-beta to activate myofibroblasts akin to the active phase of Peyronie’s disease

.jpg)
.jpg)