
Poster Session C
Rheumatoid arthritis (RA)
Daniel Toro-Domínguez, Sr, PhD
GENYO
Granada, Spain
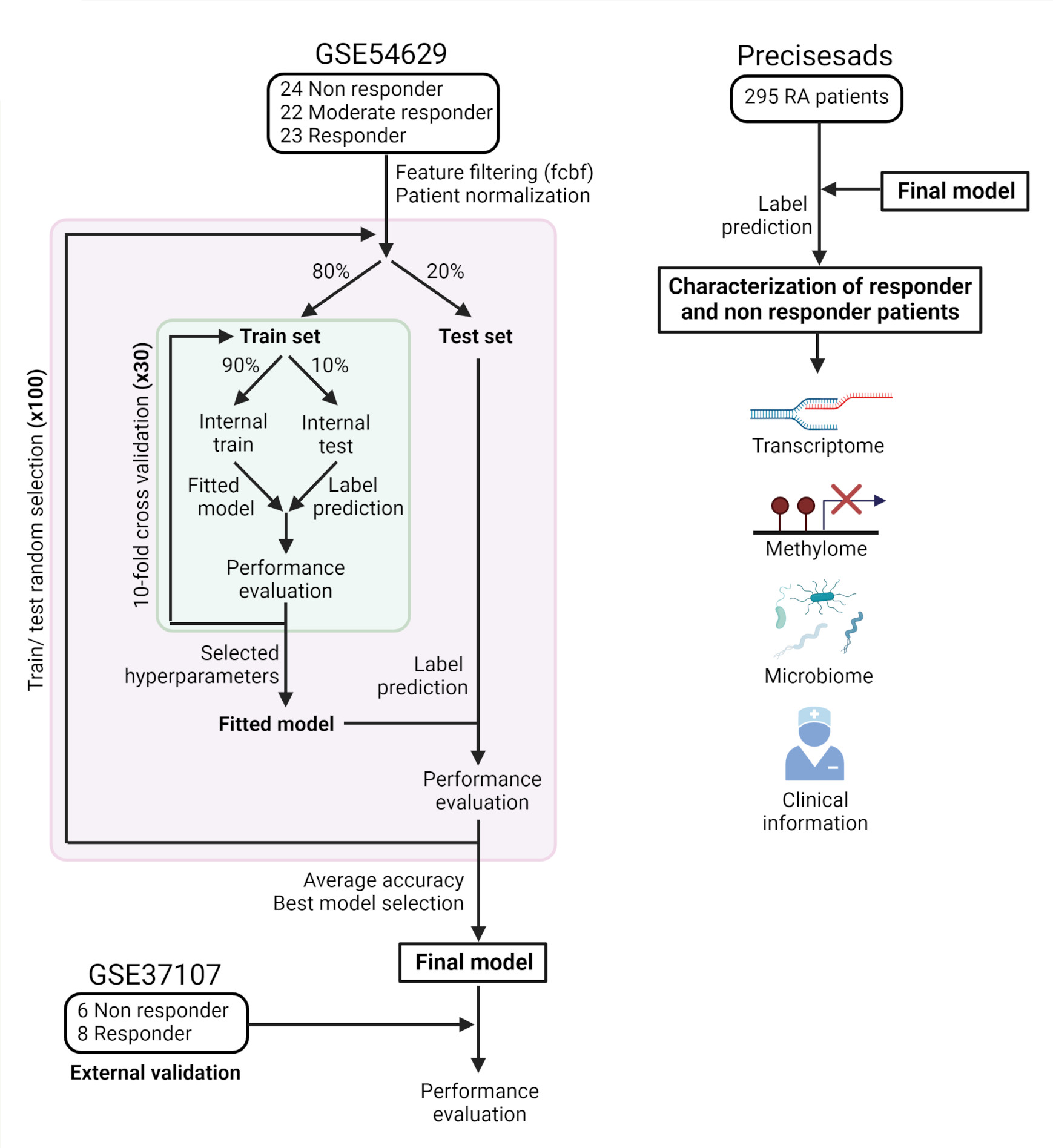 Figure 1: Summary diagram. The figure shows a diagram about the process of building the prediction model and the subsequent omic characterization of responders and non-responders patients.
Figure 1: Summary diagram. The figure shows a diagram about the process of building the prediction model and the subsequent omic characterization of responders and non-responders patients.