Princess Nourah bint Abdulrahman University Riyadh, Saudi Arabia
Fahidah Alenzi1, Shirish Sangle2 and Sanna Giovanni2, 1Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia, 2Guy's and St Thomas' NHS Foundation Trust, London, United Kingdom
Background/Purpose: Idiopathic inflammatory myositis (IIM) includes a spectrum of a rare autoimmune disease characterized by proximal muscle weakness, variable skin manifestation and extra muscular manifestations. Rituximab has been shown to be effective in refractory IIM. The aim was to describe myositis specific autoantibodies (MSA) associated clinical and laboratory characteristics including B cell depletion and ability to reduce steroid requirement in patients with IIM following rituximab treatment.
Methods: MSA associated clinical and laboratory data were retrospectively collected on IIM patients attending our Lupus Unit at Guy's and St Thomas' Trust between 2018 and 2021. We included 32 patients with refractory IIM (15 antisynthetase syndrome,17 myositis with other MSA), who received rituximab after failing to respond to multiple immunosuppressive drugs including cyclophosphamide. Numeric response variables (median and range) including age, disease duration, follow up duration, CK level pre and post treatment, albumin, inflammatory markers, immunoglobulins and degree of B cell count depletion at baseline, 6 months and 12 months following rituximab and MMT8 were collected. Frequencies and percentages categorical variables including gender, ethnicity, clinical manifestations, type of MSA, ability to reduce steroids,immunosuppression therapy used and patients' outcomes following rituximab use (clinically stable versus active) were analyzed.
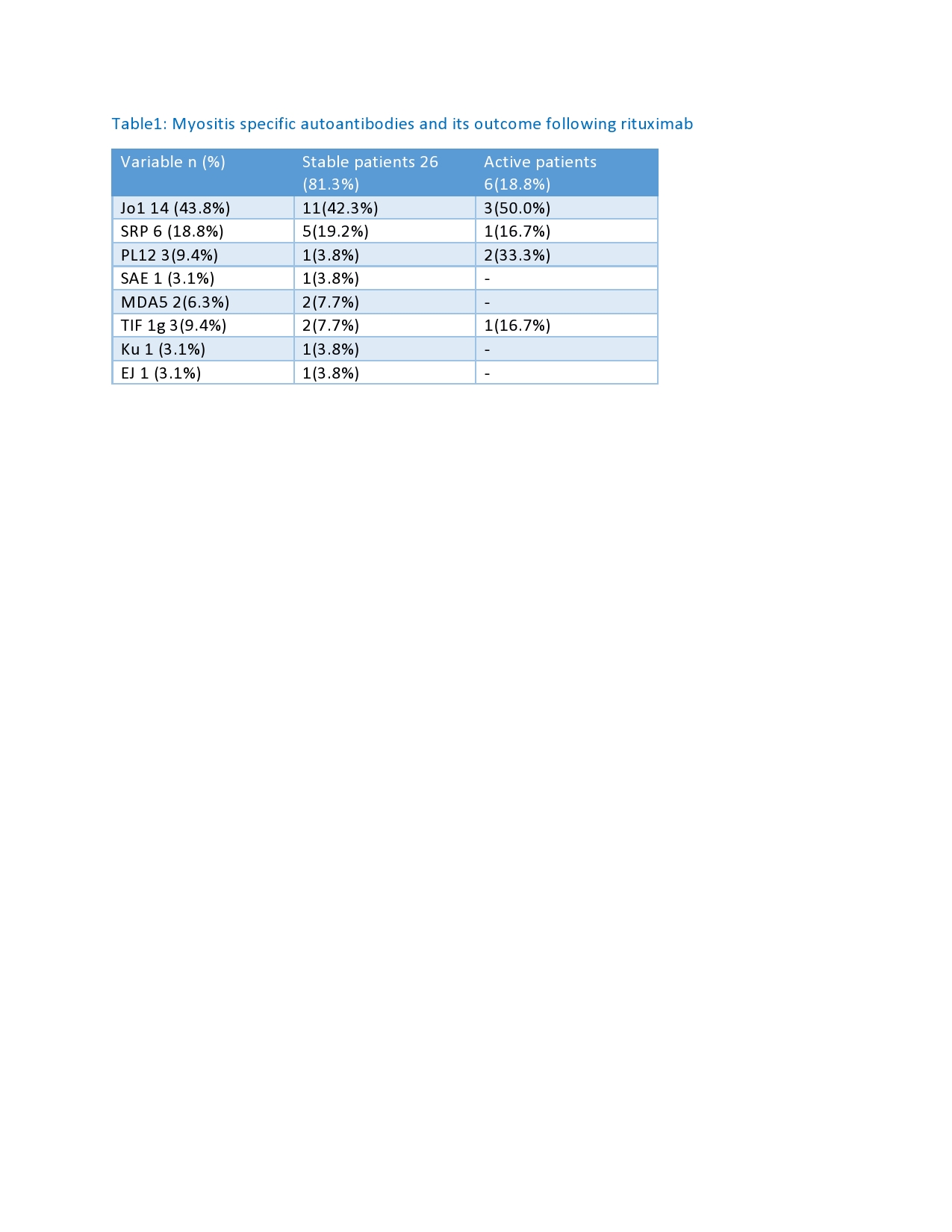
Results: 53% were female and 47% male, with a median age of 49.5 (21-72) years. Median disease duration was 6-year and median follow up 7 years. Anti-synthetase syndrome was diagnosed in 15 patients (46.9%), and 17 (53.1%) had myositis with other MSA.12 patients (37.5%) had ILD with organizing pneumonia , 8 (25%) had ILD with NSIP, 1(3.1%) had ILD with UIP, 4 (12.5%) had mixed OP-NSIP, and 3 (9.4%) had no ILD. 6 (18.8%) had cardiac involvement, 18 (56.3%) had variable skin manifestations, 3 (9.4%) had dysphagia, 26 (81.3%) arthritis, and 27 (84.4%) muscle weakness. Twenty-six patients (81.3%) responded well to treatment and achieved clinically stable disease, while 6 patients (18.8%) remained with active myositis. Out of 15 patients with anti-synthetase syndrome,11 (73.3%) reached a stable clinical outcome and 4 patients (26.7%) remained with active disease. Patients positive for anti-SAE, MDA5, Ku, or EJ, achieved a 100% response. Median CK level pre-rituximab treatment was 1612 (45-24045) IU/L, and 124.5 (35-7287) IU/L post-rituximab. Five patients (15.6%) had albumin levels less than 40 mg/dl, 4 (80%) had active disease, and 1 (20%) had a stable outcome (p< 0.01). In total, 24 (75.5%) patients were able to reduce the prednisolone < 7.5 mg daily, with 23 (95.8%) achieving a stable outcome. The median B cell count at baseline was 137.5 (9-961) cls/ml, with 1.5 (0-12) cls/ml at 6 months and 10 (0-677) cls/ml at 12 months post rituximab treatment.
Conclusion: Over 80% of our IIM patients reached clinical remission following treatment with rituximab and 75.5 % were able to significantly reduce steroid dose maintenance. Further prospective studies with larger sample sizes and longer follow-up are needed to estimate IIM outcomes after rituximab therapy. Disclosures: F. Alenzi, None; S. Sangle, None; S. Giovanni, None.