
Poster Session D
Myopathic rheumatic diseases (polymyositis, dermatomyositis, inclusion body myositis)
Sarah Tansley, PhD, BSc, MBChB
University of Bath
Bath, United Kingdom
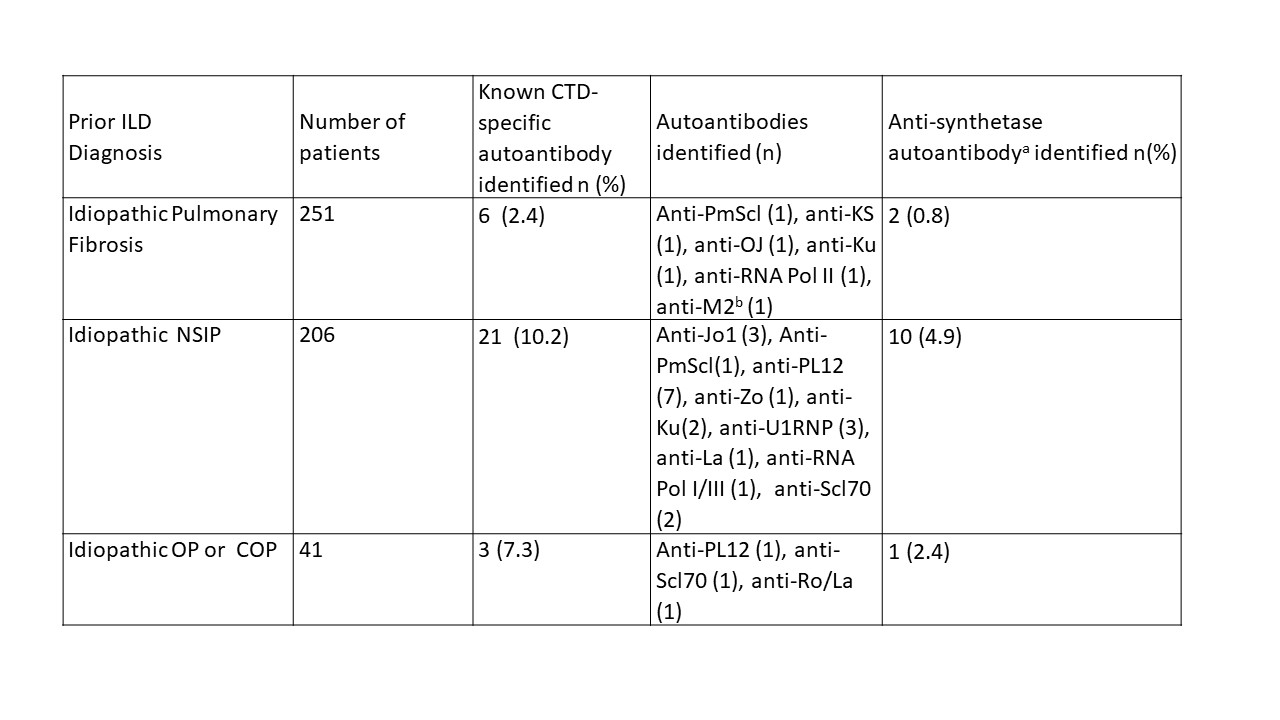 Table: The prevalence of CTD-specific autoantibodies in patients with a diagnosis of idiopathic interstitial lung disease
Table: The prevalence of CTD-specific autoantibodies in patients with a diagnosis of idiopathic interstitial lung disease