
Ignite Talk
Liam O'Neil, MD, MHS
University of Manitoba
Winnipeg, MB, Canada
Disclosure: Disclosure information not submitted.
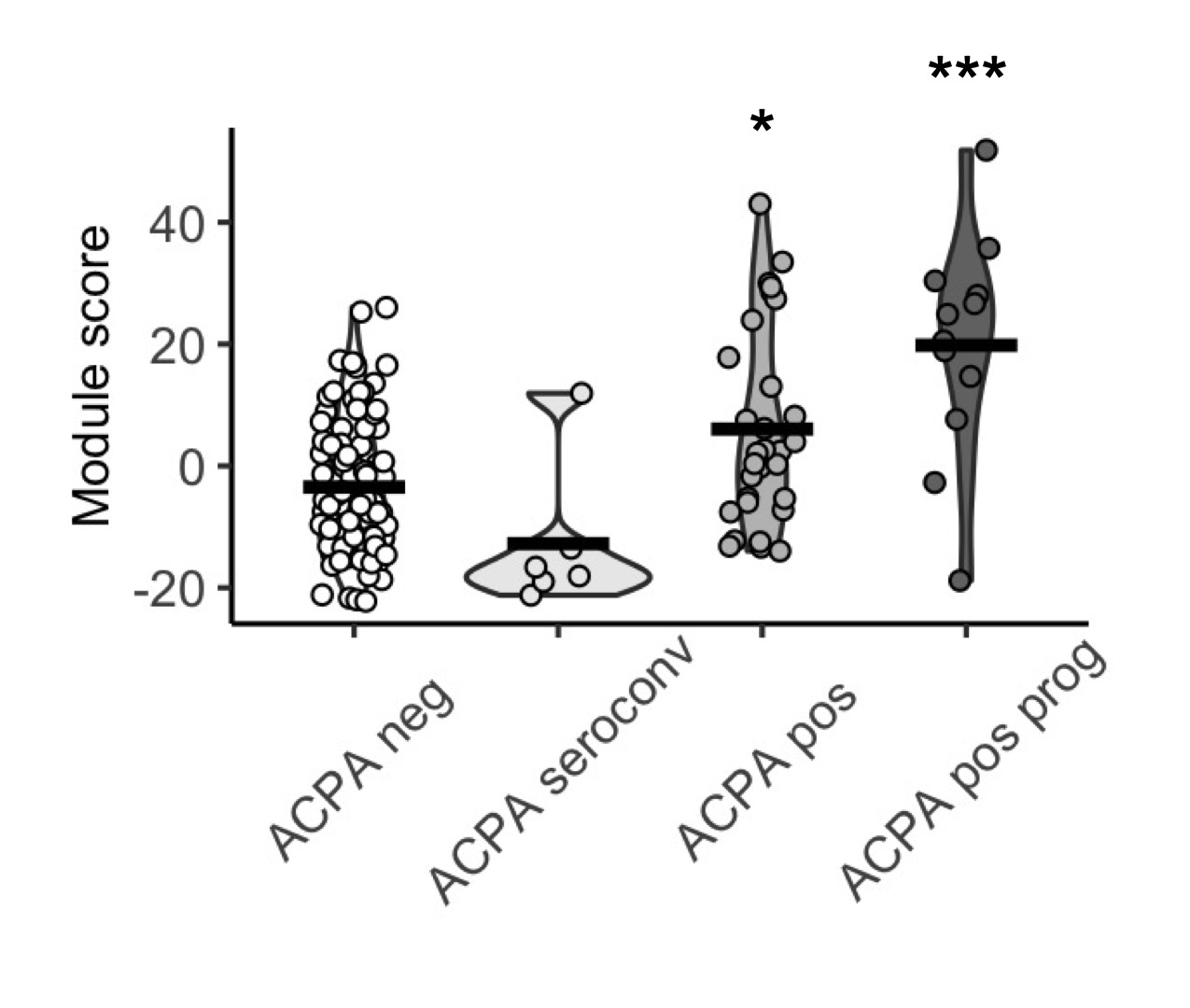 A serum protein module score measured in baseline serum samples is associated with progression into inflammatory arthritis in FDR of RA patients. The score was not significantly different between ACPA FDR who underwent seroconversion and ACPA negative FDR.
A serum protein module score measured in baseline serum samples is associated with progression into inflammatory arthritis in FDR of RA patients. The score was not significantly different between ACPA FDR who underwent seroconversion and ACPA negative FDR.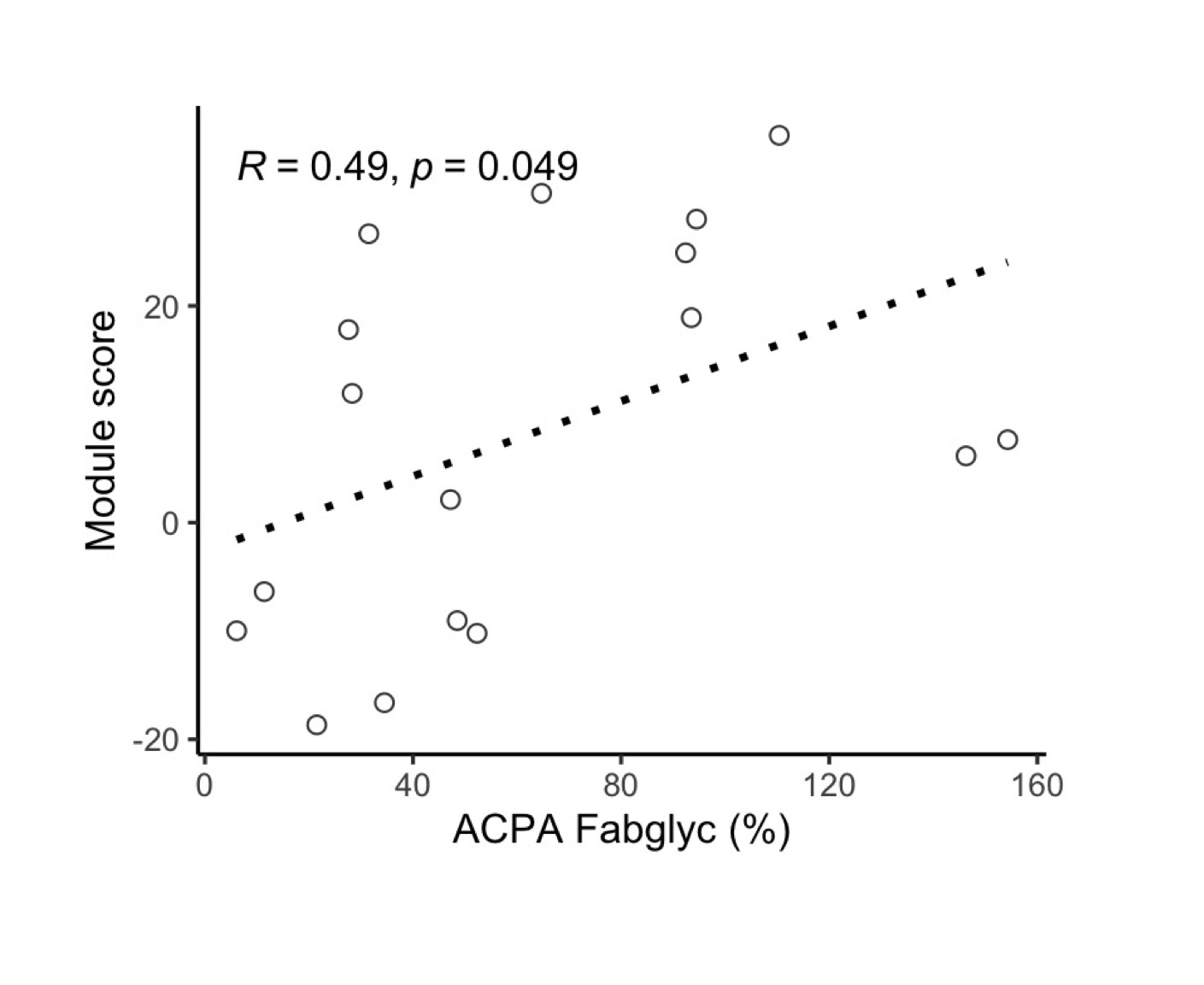 A serum protein module score correlated with ACPA IgG Fab glycosylation.
A serum protein module score correlated with ACPA IgG Fab glycosylation.