Paul Newcombe1, Richard A. Furie2, Yoshiya Tanaka3, Wendy White4, Dominic Sinibaldi4, Philip Brohawn4, Mark Lazarus1, Nicola Ferrari1, Raj Tummala4, Hussein Al-Mossawi1, Andre Nogueria da Costa5, Daniel Muthas5 and Madhu Ramaswamy4, 1AstraZeneca, Cambridge, United Kingdom, 2Northwell Health, Great Neck, NY, 3University of Occupational and Environmental Health, Kitakyusyu Fukuoka, Japan, 4AstraZeneca, Gaithersburg, MD, 5AstraZeneca, Gothenburg, Sweden
Background/Purpose: SLE has been associated with expression of type I IFN gene signatures (IFNGS).1 Anifrolumab, a monoclonal antibody binding IFN receptor subunit 1, inhibits downstream type I IFN signaling. Anifrolumab treatment has resulted in favorable clinical outcomes (including BICLA and CLASI responses and oral glucocorticoid [GC] tapers) in adult patients with moderate to severe SLE despite standard therapy in 2 phase 3 trials, TULIP-1 and TULIP-2.2,3 The objective was to identify pharmacodynamic (PD) biomarkers associated with anifrolumab vs placebo treatment using proteomic assessments of longitudinal plasma samples from TULIP-1.
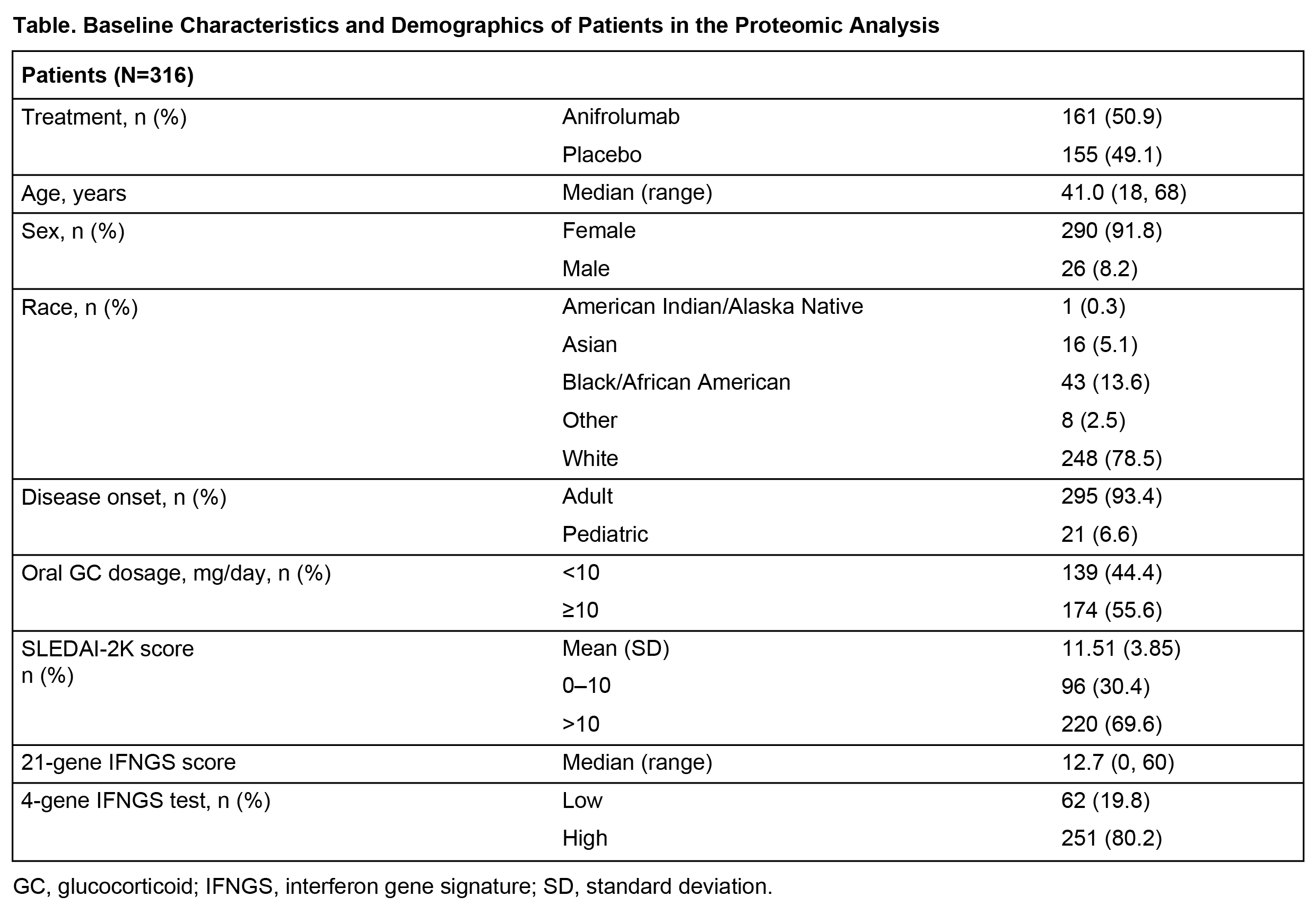
Methods: Plasma samples from 316 patients fulfilling the 1997 ACR criteria for SLE in TULIP-1 were assessed for 96 proteins using multiplexed Luminex assays (86 analytes) and Simoa technology (n=10). Of 96 proteins assayed, 85 were ≥ the lower limit of quantitation in ≥50% of samples; 4 were removed from analysis due to significant baseline imbalances across treatment allocations. Principal component analysis indicated 4 sample outliers, which were removed from further analyses. A longitudinal mixed effect model was used to test whether 1-year trajectories of protein levels were statistically different between anifrolumab and placebo groups. Multiple testing of 81 proteins was accounted for using the Benjamini-Hochberg False Discovery Rate (FDR) procedure (0.05 threshold); analyses were adjusted for SLEDAI-2K, oral GC dose, age, sex, race, and BMI.
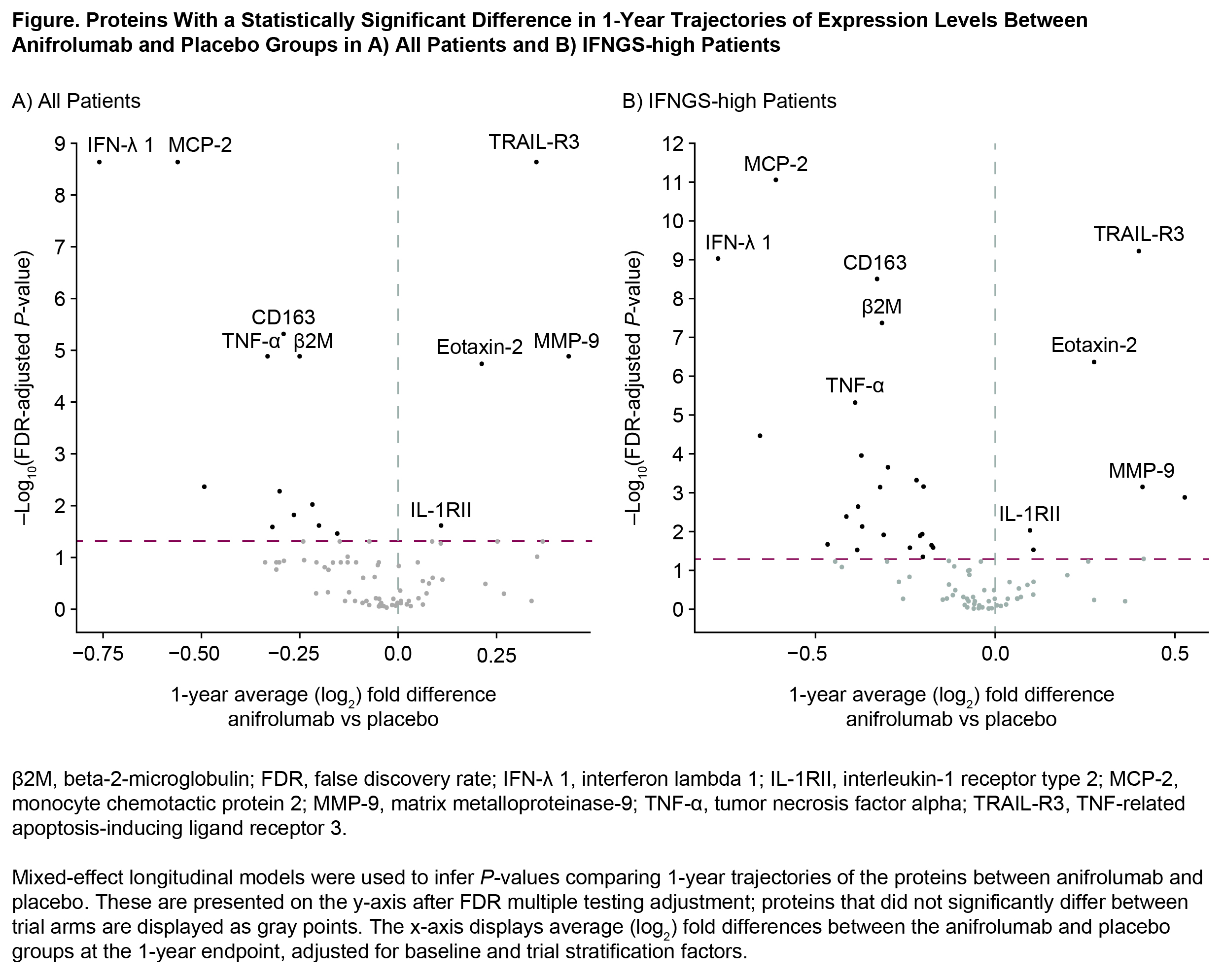
Results: Patient demographics and baseline characteristics are shown in Table. Proteomic analysis revealed 36 of 81 proteins statistically associated with high IFNGS at baseline. Of these 36, 33 proteins were upregulated in IFNGS-high vs IFNGS-low patients. Longitudinal assessment of 1-year trajectories of 81 protein levels identified 16 proteins differentially impacted by anifrolumab vs placebo treatment (Figure, A). Proteins most strongly influenced by anifrolumab exposure included MCP-2, TRAIL-R3 (all FDR < 1e-8), and CD163, TNF-α, β2M, MMP‑9, and Eotaxin-2 (all FDR < 1e-4). In IFNGS-high patients, anifrolumab treatment was associated with larger numbers of proteins (n=29) with significantly different trajectories vs placebo (Figure, B); however, the most significantly impacted proteins were the same as in the overall population. Observed proteomic PD impact of anifrolumab corresponds to multimodal type I IFN pathway effects, including suppression of T/B cell cytokines (BAFF, IFN-γ), innate cell chemokines and receptors (MCP-2, Eotaxin-2), and upregulation of antiapoptotic factors (TRAIL-R3, IL-1RII); this expands and corroborates results from an analysis of samples from the phase 2 anifrolumab study.4
Conclusion: We provide evidence that anifrolumab significantly suppresses type I IFN-stimulated proteins involved in T, B, and innate cell activation and chemotaxis. Our results contribute to a deeper understanding of how anifrolumab affects SLE disease activity.
References: 1. Crow MK. Curr Opin Rheumatol. 2014;26:467–74. 2. Furie R. Lancet Rheumatol. 2019;1:e208–19. 3. Morand EF. N Engl J Med. 2020;382:211–21. 4. Casey KA. Lupus Sci Med. 2018;5:e000286.
Disclosures: P. Newcombe, AstraZeneca; R. Furie, AstraZeneca, Biogen; Y. Tanaka, Behringer Ingelheim, Eli Lilly, Abbvie, Gilead, AstraZeneca, Bristol-Myers, Chugai, Daiichi-Sankyo, Eisai, Pfizer, Mitsubishi-Tanabe, GlaxoSmithKline, Asahi-Kasei; W. White, AstraZeneca; D. Sinibaldi, AstraZeneca, Neuraly; P. Brohawn, AstraZeneca; M. Lazarus, AstraZeneca, Novartis; N. Ferrari, AstraZeneca; R. Tummala, AstraZeneca; H. Al-Mossawi, AstraZeneca; A. Nogueria da Costa, AstraZeneca; D. Muthas, AstraZeneca; M. Ramaswamy, AstraZeneca.