
Abstract Session
Fibrosing rheumatic diseases (scleroderma, MCTD, IgG4-related disease, scleroderma mimics)
.png "Benjamin D. Korman, MD photo")
Benjamin D. Korman, MD
University of Rochester
Rochester, NY, United States
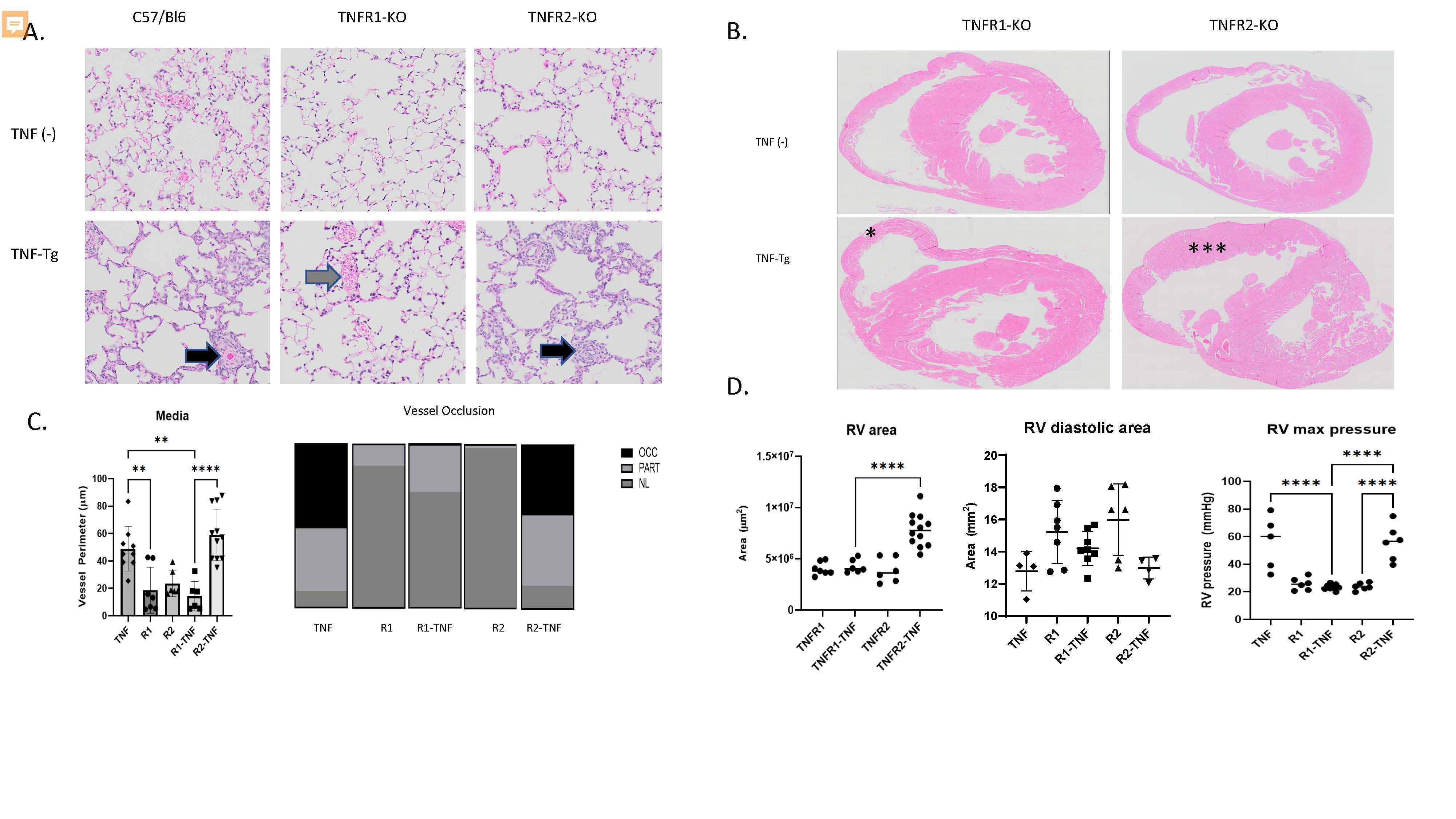 Figure 1. TNFR1 mediates PAH and arthritis phenotype in a mouse model of CTD-PAH. A. Lung histology (H&E, 20x) images demonstrate that TNF-Tg and TNFR2-KO/TNF-Tg mice, but not TNFR1-KO/TNF-Tg mice have pulmonary inflammation and vascular occlusion (arrowheads). B. Cardiac histology (H&E, 4x) demonstrates RV hypertrophy in TNFR2-KO/TNF-Tg (**) mice but not TNFR1-KO/TNF-Tg mice (*). C. (Left) Quantification of pulmonary vessel media perimeter (Right) Proportion of partially and fully occluded vessels in each phenotype. D. (Left) RV area measured by histomorphometry, (Middle) RV end diastolic area measured by echocardiography (Right) RV systolic pressure measured by right heart catheterization.
Figure 1. TNFR1 mediates PAH and arthritis phenotype in a mouse model of CTD-PAH. A. Lung histology (H&E, 20x) images demonstrate that TNF-Tg and TNFR2-KO/TNF-Tg mice, but not TNFR1-KO/TNF-Tg mice have pulmonary inflammation and vascular occlusion (arrowheads). B. Cardiac histology (H&E, 4x) demonstrates RV hypertrophy in TNFR2-KO/TNF-Tg (**) mice but not TNFR1-KO/TNF-Tg mice (*). C. (Left) Quantification of pulmonary vessel media perimeter (Right) Proportion of partially and fully occluded vessels in each phenotype. D. (Left) RV area measured by histomorphometry, (Middle) RV end diastolic area measured by echocardiography (Right) RV systolic pressure measured by right heart catheterization. 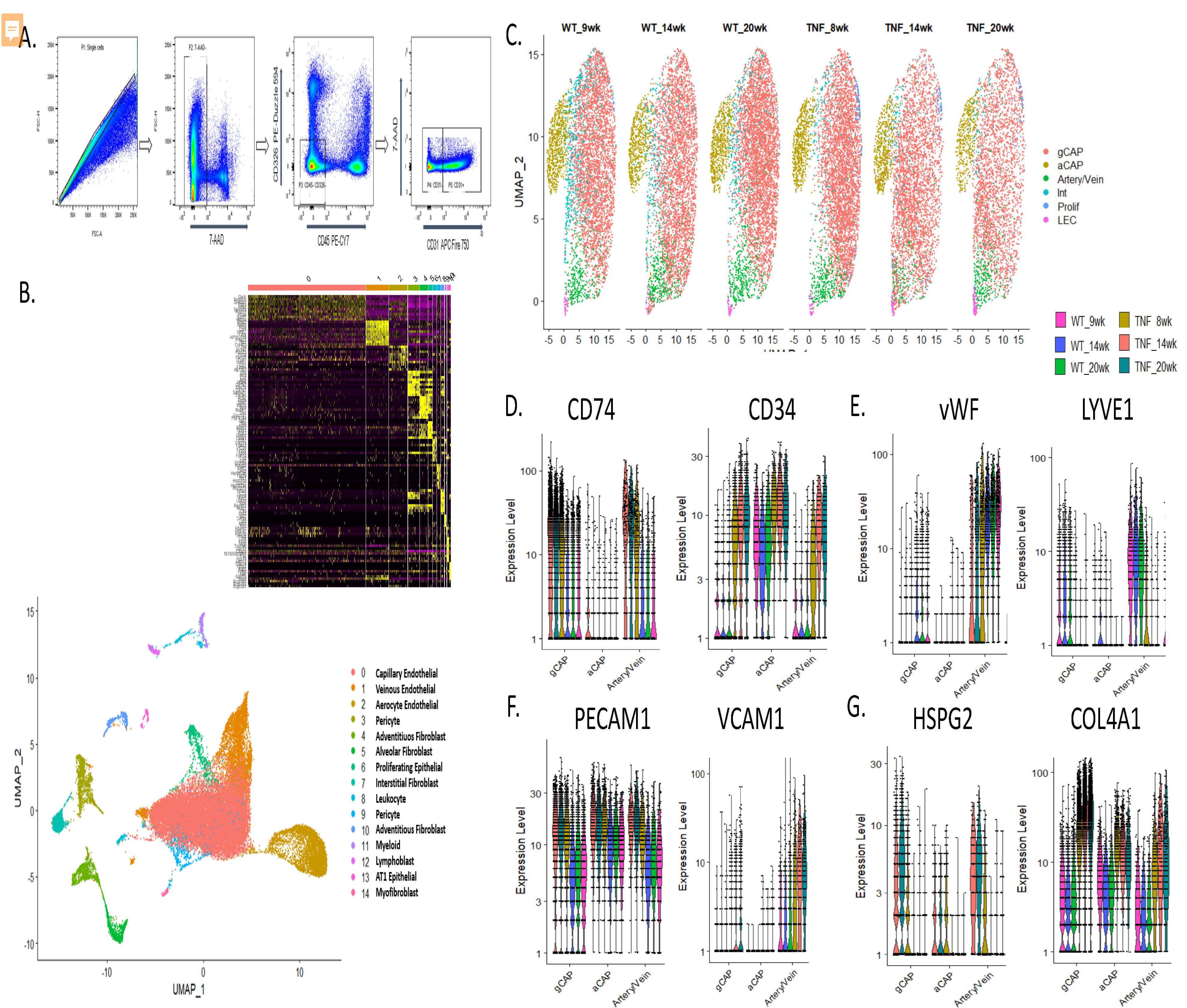 Figure 2. Single cell digestion, sorting, and characterization of endothelial cell phenotypes in TNF-Tg lungs. A. Flow cytometric gating strategy to deplete lungs of CD45+ and CD326+ cells. B. uMAP projection demonstrating 15 pulmonary endothelial and menenchymal cellular populations identified after single cell RNA sequencing across 43,455 cells sequenced. (inset upper) heatmap demonstrating canonical genes used to identify cellular sub-populations C. uMAP projection demonstrating subsets of endothelial cells, note decreased cellular density of general capillary endothelial cells (gCAP) particularly at the 14 and 20 week timepoints in TNF-Tg mice. D. Up-regulation of CD34 and down-regulation of CD74, markers previously shown to be differentially regulated in human PAH, in TNF-Tg endothelial cells. E. Loss of canonical venous and arterial markers vWF and up-regulation of LYVE1 in arterial /venous endothelial cells. F. Over-expression of adhesion molecules PECAM1 and loss of VCAM1 in endothelial cells. G. Over-expression of basement membrane components HSPG2 and Col4a1 across multiple endothelial cell types
Figure 2. Single cell digestion, sorting, and characterization of endothelial cell phenotypes in TNF-Tg lungs. A. Flow cytometric gating strategy to deplete lungs of CD45+ and CD326+ cells. B. uMAP projection demonstrating 15 pulmonary endothelial and menenchymal cellular populations identified after single cell RNA sequencing across 43,455 cells sequenced. (inset upper) heatmap demonstrating canonical genes used to identify cellular sub-populations C. uMAP projection demonstrating subsets of endothelial cells, note decreased cellular density of general capillary endothelial cells (gCAP) particularly at the 14 and 20 week timepoints in TNF-Tg mice. D. Up-regulation of CD34 and down-regulation of CD74, markers previously shown to be differentially regulated in human PAH, in TNF-Tg endothelial cells. E. Loss of canonical venous and arterial markers vWF and up-regulation of LYVE1 in arterial /venous endothelial cells. F. Over-expression of adhesion molecules PECAM1 and loss of VCAM1 in endothelial cells. G. Over-expression of basement membrane components HSPG2 and Col4a1 across multiple endothelial cell types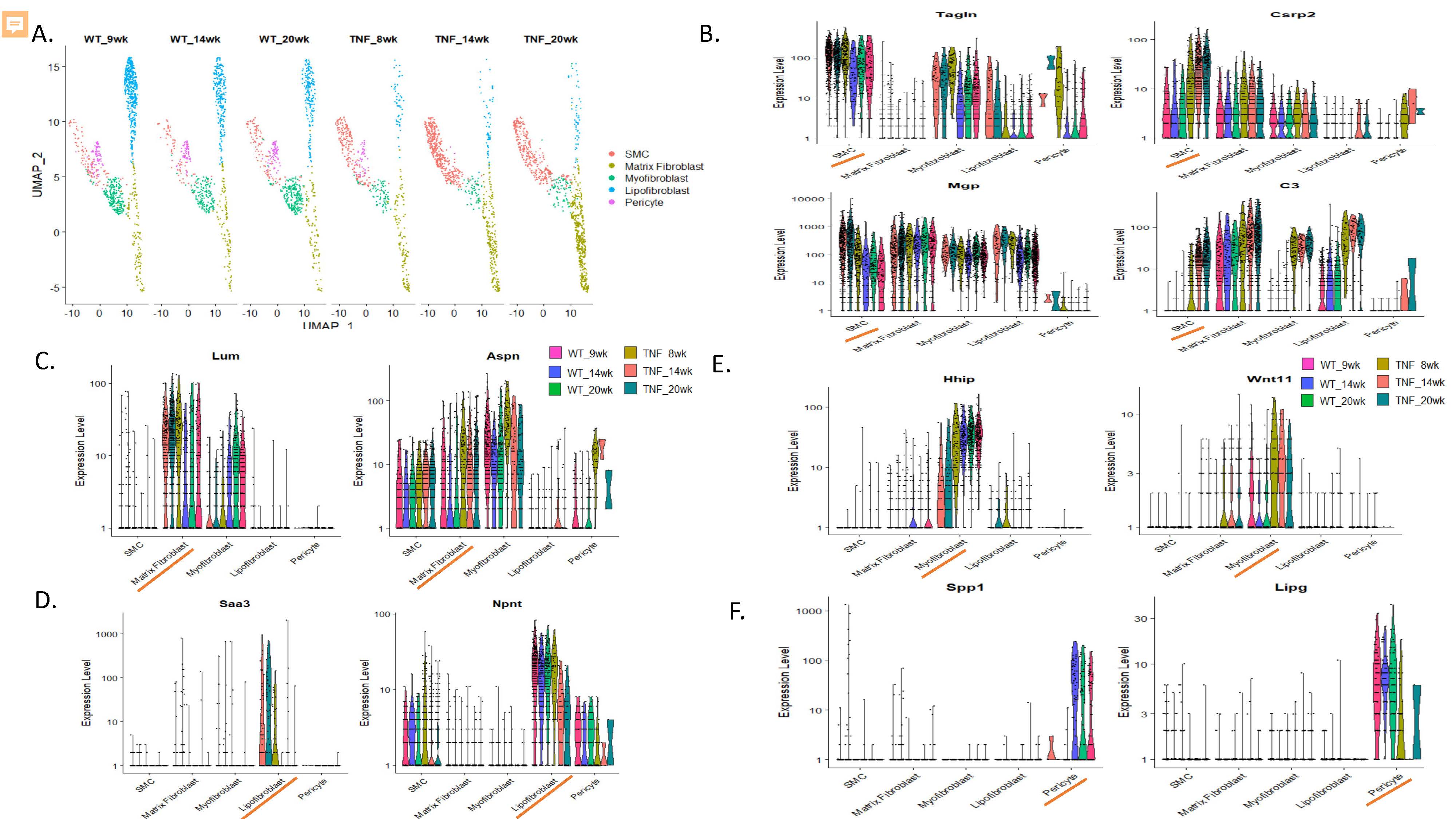 Figure 3. Alterations in mesenchymal cell types in TNF-Tg lungs. A. uMAP projection demonstrating subsets of mesenchymal cells, note increases in smooth muscle cells and matrix fibroblast populations and decreases in pericytes, lipofibroblasts, and myofibroblasts in TNF-Tg mice. C. Over-expression of TAGLN, CSRP2, MGP, and C3 in TNF-Tg smooth muscle cells. D. Over-expression of LUM and DCN in matrix fibroblasts. D. Loss of NPNT and over-expression of SAA3 in TNF-Tg lipofibroblasts. E. Loss of HHIP and over-expression of WNT11 in TNF-Tg myofibroblasts. F. Loss of SPP1 and LIPG expression in TNF-Tg lung pericytes
Figure 3. Alterations in mesenchymal cell types in TNF-Tg lungs. A. uMAP projection demonstrating subsets of mesenchymal cells, note increases in smooth muscle cells and matrix fibroblast populations and decreases in pericytes, lipofibroblasts, and myofibroblasts in TNF-Tg mice. C. Over-expression of TAGLN, CSRP2, MGP, and C3 in TNF-Tg smooth muscle cells. D. Over-expression of LUM and DCN in matrix fibroblasts. D. Loss of NPNT and over-expression of SAA3 in TNF-Tg lipofibroblasts. E. Loss of HHIP and over-expression of WNT11 in TNF-Tg myofibroblasts. F. Loss of SPP1 and LIPG expression in TNF-Tg lung pericytes