
Abstract Session
Fibrosing rheumatic diseases (scleroderma, MCTD, IgG4-related disease, scleroderma mimics)

Julie Paik, MD, MHS
Johns Hopkins University
Baltimore, MD, United States
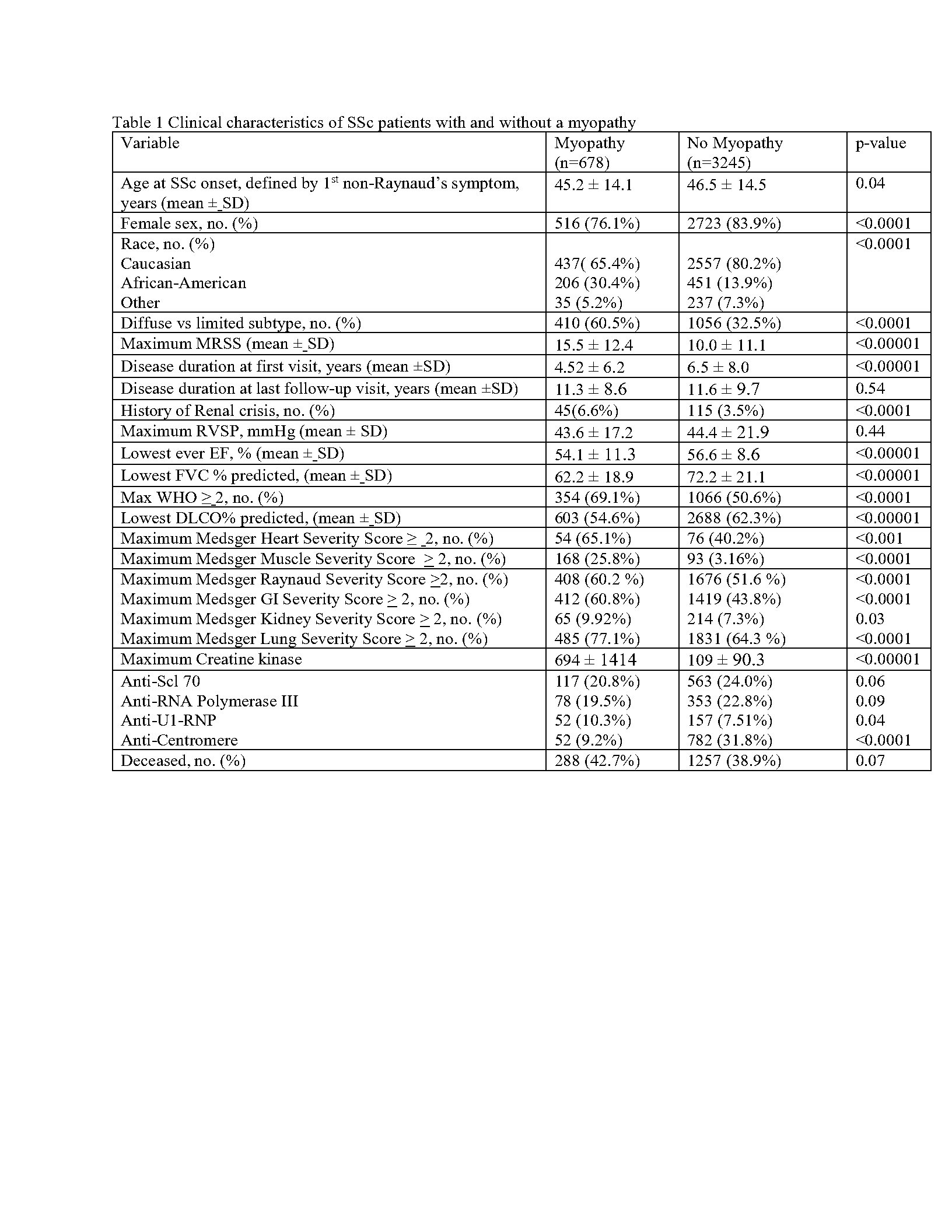
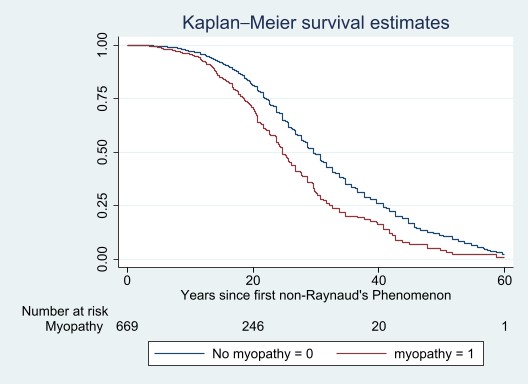 Kaplan Meier survival estimates comparing scleroderma patients who have myopathy versus those who do not have myopathy and time till death
Kaplan Meier survival estimates comparing scleroderma patients who have myopathy versus those who do not have myopathy and time till death