
Poster Session A
Spondyloarthritis (SpA) including psoriatic arthritis (PsA)
Mark C. Hwang, MD, MS
McGovern Medical School at the University of Texas Health Science Center at Houston
Houston, TX, United States
.jpg)
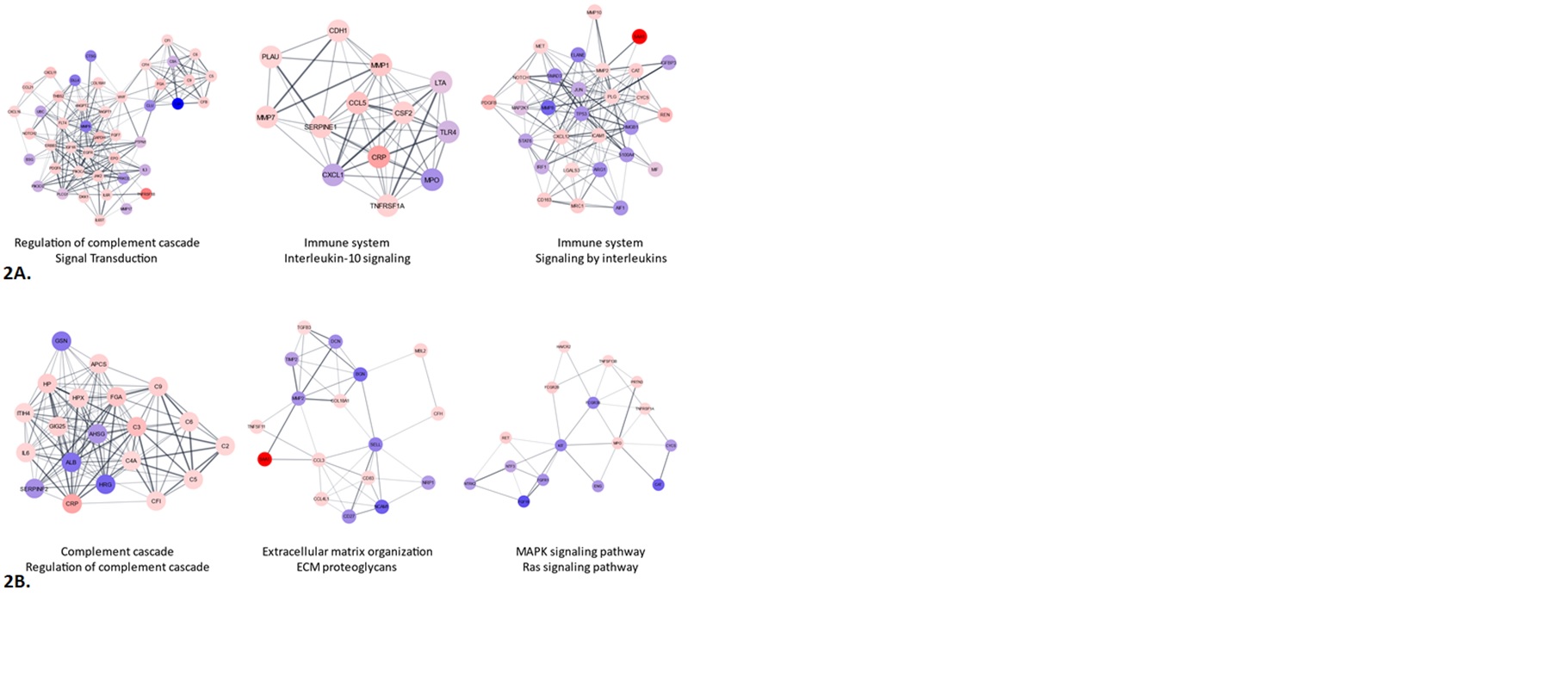 Figure 2. Protein-protein interaction networks 2A. Top 367 Diagnosis DEPs and 2B. Top 157 Monitoring DEPs (T-test p < 0.05) through the Cytoscape string App with a confidence cutoff of 0.4. MCODE clustering was performed and displayed are the top three clusters. The color of each node corresponds to the fold change. Nodes with a fold change less than one range in color from blue to purple while those with a fold change greater than one range from pink to red. The confidence score of each interaction is displayed as the edge thickness and opacity. The top two reactome pathways associated with each cluster are displayed below each cluster.
Figure 2. Protein-protein interaction networks 2A. Top 367 Diagnosis DEPs and 2B. Top 157 Monitoring DEPs (T-test p < 0.05) through the Cytoscape string App with a confidence cutoff of 0.4. MCODE clustering was performed and displayed are the top three clusters. The color of each node corresponds to the fold change. Nodes with a fold change less than one range in color from blue to purple while those with a fold change greater than one range from pink to red. The confidence score of each interaction is displayed as the edge thickness and opacity. The top two reactome pathways associated with each cluster are displayed below each cluster.