0037: Dual (and Dueling) Fibroblast Differentiation Is Integral to the Pathogenesis of Anti-Ro Mediated Congenital Heart Block and Provides a Novel Biomarker of Severity
NYU Grossman School of Medicine Brooklyn, NY, United States
Christina Firl1, Robert Clancy2, Bettina Cuneo3, Nicola Fraser4, Marc Halushka5, Mala Masson6, Amit Saxena1 and Jill Buyon2, 1NYU Langone Health, New York, NY, 2NYU Grossman School of Medicine, New York, NY, 3University of Colorado School of Medicine, Aurora, CO, 4NYU School of Medicine, New York, NY, 5Johns Hopkins Medicine, Baltimore, MD, 6NYU Langone Medical Center- Division of Rheumatology, New York, NY
Background/Purpose: Fibroblast heterogeneity may provide critical insights into autoimmune mediated tissue damage. Transdifferentiation to a scarring myofibroblast is characterized by expression of alpha-Smooth Muscle Actin (α-SMA), deposition of collagen and contractile properties while another fibroblast subtype evaluated largely in tumor migration is distinguished by expression of S100 Calcium Binding Protein A4 (S100A4). Given the histologic hallmark of AV nodal fibrosis and endocardial fibroelastosis in anti-Ro mediated cardiac neonatal lupus (NL), this study addressed the contribution of S100A4 and α-SMA-expressing fibroblasts.
Methods: In an in vitro model of cardiac-NL, 2nd trimester human fetal cardiac fibroblasts were co-cultured with supernatants (sup) of macrophage-like THP-1 cells transfected with hY3 (noncoding ssRNA TLR7/8 agonist binding Ro60) and additional treatments with readouts: immunofluorescence, flow cytometry, qPCR, and a collagen assay. S100A4 was measured in anti-Ro exposed cord bloods and expression evaluated in fetal cardiac tissue.
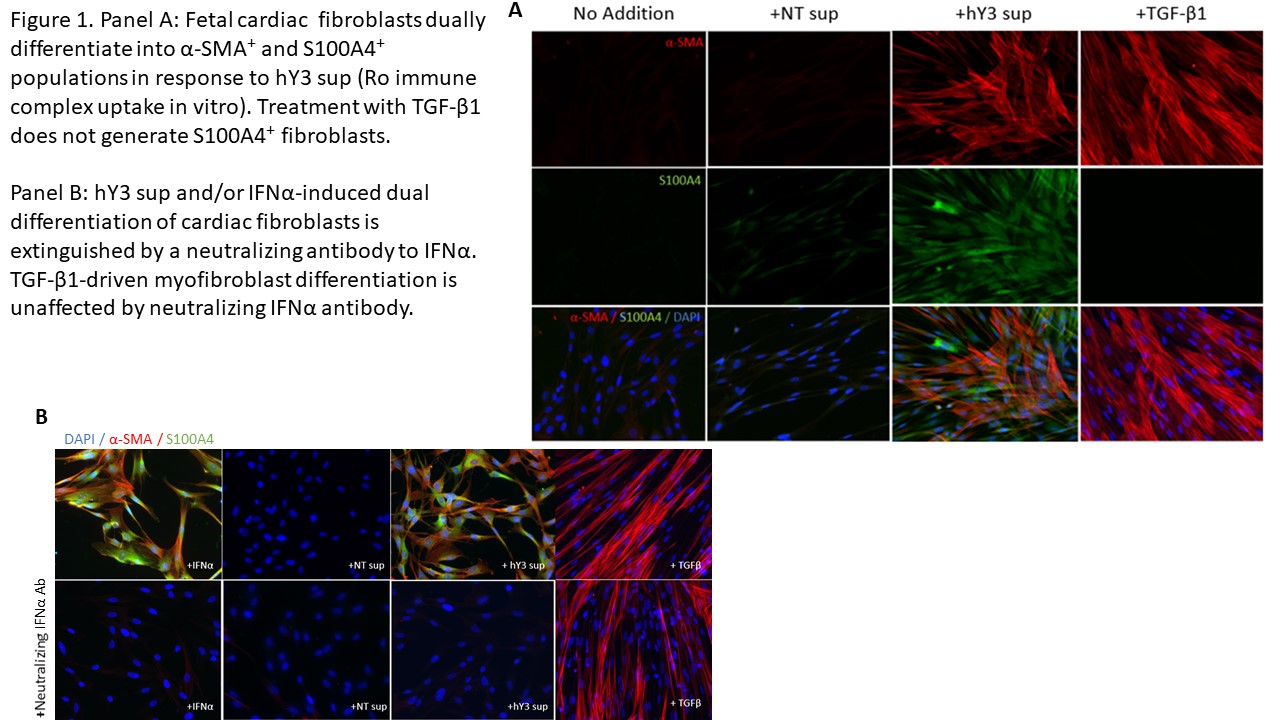
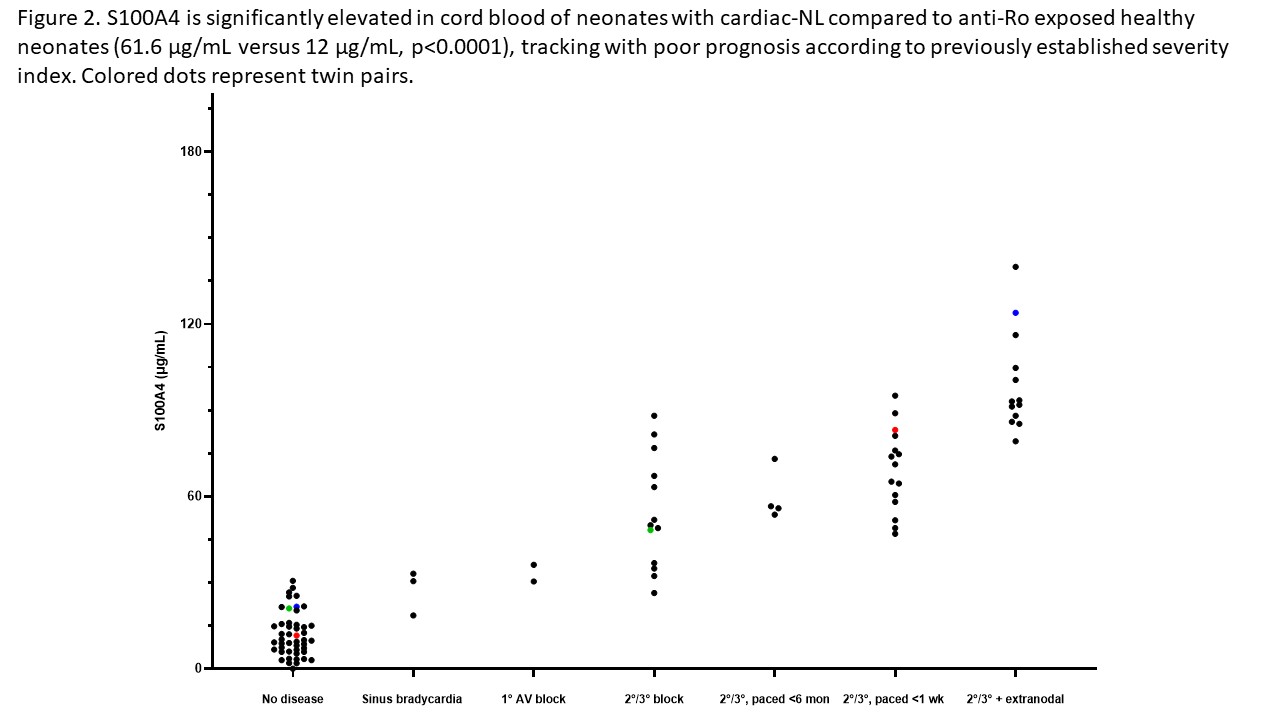
Results: When cultured with hY3 sup, cardiac fibroblasts differentiated into two populations, distinguished by expression of α-SMA or S100A4. In contrast, activated TGF-β1 only resulted in expression of α-SMA (Figure 1). These results were confirmed by flow cytometry; hY3 sup promoted expression of both α-SMA (8263±5185) and S100A4 (1316±469) whereas TGF-β1 stimulated fibroblasts expressed α-SMA (9085±5867) not S100A4 (159.2±56.9) (p=0.002, N=4). Unlike TGF-β1 but similar to hY3 sup, IFNα induced dual fibroblast differentiation (α-SMA-16332±9085, S100A4-1698±806.4, p=0.008, N=3). Dual differentiation induced by hY3 sup or IFNα was inhibited by anti-IFNα which had no effect on TGF-β1-induced α-SMA. hY3-treated fibroblasts upregulated transcription of proangiogenic cytokines IL6, IL10 and MMP13 (ECM remodeling) compared to TGF-β1 (5.7 vs 1.5, p=0.0079, N=5; 5.1 vs 0.96, p=0.032, N=3; 7.5 vs 0.8, p=0.0079, N=5) while expression indicative of collagen deposition (Col1α1) was upregulated by TGF-β1 compared to hY3 sup (4.8 vs 1.8, p=0.028, N=4). In distinction, S100A4-positive fibroblasts uniquely degraded soluble collagen (14 ug/mL to 0.6, p=0.026, N=5). Bolstering in vivo relevance, circulating S100A4 was elevated in cardiac-NL (N=49) compared to non-cardiac-NL (N=47) (61.6 vs 12.0 ug/mL, p< 0.0001). Remarkably, levels tracked with an established cardiac severity score and in twins, distinguished the affected from the unaffected neonate (Figure 2). S100A4 immunostaining was observed in 3 cardiac-NL hearts focally in fibroblasts within the conducting tissue but not in a healthy heart. α-SMA was detected in fibrotic zones while S100A4 in regions with dystrophic calcification.
Conclusion: In vitro and in vivo experiments position the S100A4-positive fibroblast subtype alongside the canonical myofibroblast in the pathogenesis of cardiac-NL. The pro-angiogenic and distinct collagenase activity may represent a thwarted attempt to mitigate fibrotic sequelae by the collagen-secreting myofibroblast with resultant unintended soft tissue calcification. Moreover neonatal S100A4 levels support a novel biomarker of poor prognosis.
Disclosures: C. Firl, None; R. Clancy, None; B. Cuneo, None; N. Fraser, None; M. Halushka, None; M. Masson, None; A. Saxena, None; J. Buyon, Equillium, GlaxoSmithKlein(GSK), L and M Healthcare Communications, Janssen, Boomcom, Merck/MSD.