University of Cincinnati Medical Center Cincinnati, OH
Debra Yen, MD1, Divya Sharma, MD1, Marshall Weesner, MD2, Michael Schoech, MD1 1University of Cincinnati Medical Center, Cincinnati, OH; 2Cincinnati VA Medical Center, Cincinnati, OH
Introduction: Primary AL amyloidosis is a systemic disease characterized by the deposition of insoluble fibrils from immunoglobulin light chains. Hepatic involvement is common, and portal hypertension, jaundice, and liver failure have been described. We present a case of primary hepatic amyloidosis that mimicked the rapid development of decompensated cirrhosis.
Case Description/Methods: A 63-year-old male with a history of remotely treated hepatitis C presented with new onset ascites over one month. Laboratory values were notable for AST 201, ALT 64, TBili 1.3, ALP 582, albumin 2.5, platelet count 105, INR 1.5. Paracentesis was performed with 4 liters removed. Serum ascites albumin gradient (SAAG) was > 1.1, protein < 2.0. Other features of his presentation included acute kidney injury with creatinine 3.1 and nephrotic range proteinuria. Of note, he had a liver biopsy 5 years ago with only portal fibrosis. Serologic evaluation for causes of chronic liver disease was negative. Further testing revealed monoclonal free kappa light chains on urine immunofixation and an elevated kappa/lambda ratio of 8.35. Both liver and kidney biopsies displayed amyloid confirmed by Congo red stain, and he was diagnosed with AL amyloidosis. However, a week later he expired from ventricular fibrillation cardiac arrest.
Discussion: This case highlights the diagnostic dilemma of a patient without known liver disease who rapidly developed signs of portal hypertension and systemic features of other end-organ involvement (i.e. proteinuria, cardiomyopathy), demonstrating the importance of including amyloidosis in the differential. In this case, the patient had a recent liver biopsy that was unremarkable. On presentation, however, he seemed to have progressed to decompensated cirrhosis in a short period of time, with ascites presumed to be from portal hypertension given the elevated SAAG. Ultimately, his entire presentation was attributable to primary hepatic amyloidosis.
While clinical manifestations of hepatic amyloidosis are uncommon, with hepatomegaly and elevated alkaline phosphatase as the most frequent findings, sinusoidal portal hypertension may occur and manifest as ascites, splenomegaly, and bleeding esophageal varices. The portal hypertension is thought to be related to the decreased vascular space of hepatic sinusoids from massive perisinusoidal amyloid deposits. Diagnosis involves biopsy of other organs and/or the liver, in which bleeding is a risk. Prognosis is poor for hepatic amyloidosis, with median survival of 9 months.
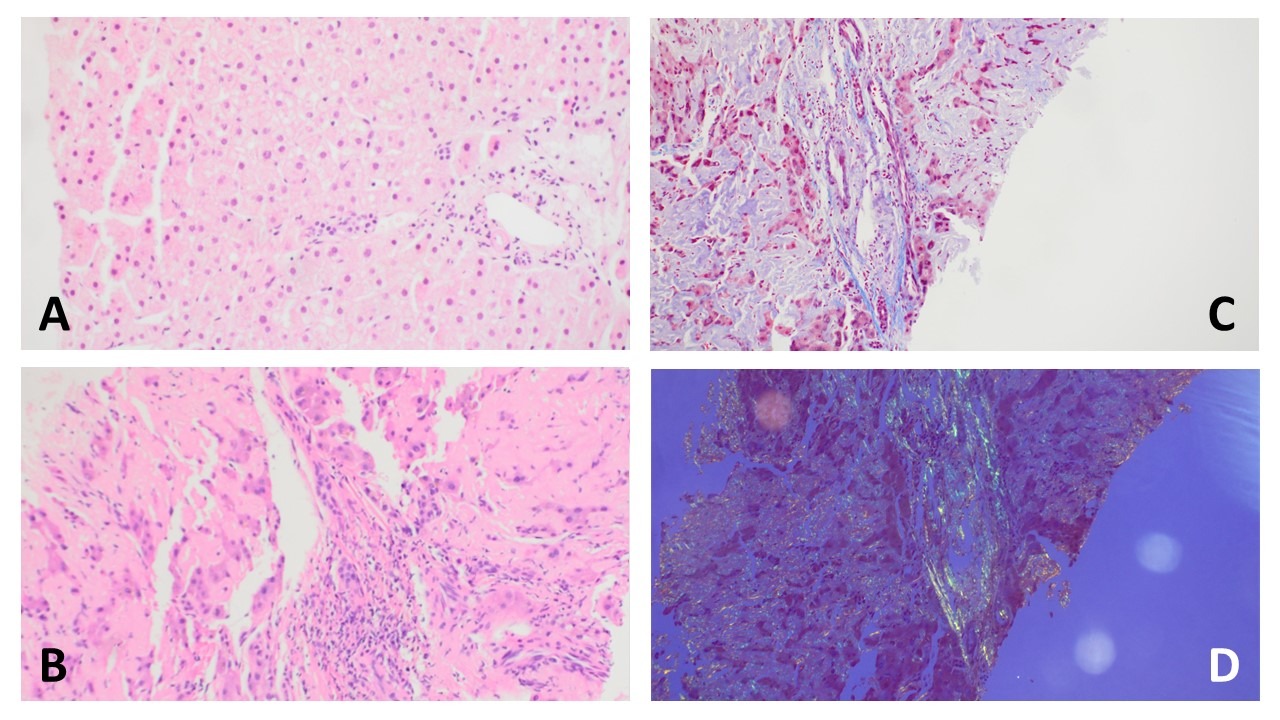
Figure: A. H&E stain 20X (2017): Normal hepatic parenchymal architecture. No significant fibrosis, inflammation or amyloid deposition. B. H&E stain 10X (2022): Deposition of abundant amorphous, acellular, eosinophilic amyloid deposits within portal vessels and hepatic sinusoids leading to hepatocyte atrophy. C. Trichrome stain 20X (2022): gray blue amyloid deposits within sinusoidal spaces and within portal tracts. Bright blue collagen deposition is minimal. D. Congo red stain 10X (2022): Apple-green birefringence under polarized light within sinusoids and portal tracts.
Disclosures:
Debra Yen indicated no relevant financial relationships.
Divya Sharma indicated no relevant financial relationships.
Marshall Weesner indicated no relevant financial relationships.
Michael Schoech indicated no relevant financial relationships.
Debra Yen, MD1, Divya Sharma, MD1, Marshall Weesner, MD2, Michael Schoech, MD1. E0573 - Primary Hepatic Amyloidosis as a Mimic of the Rapid Development of Decompensated Cirrhosis, ACG 2022 Annual Scientific Meeting Abstracts. Charlotte, NC: American College of Gastroenterology.