Medical College of Wisconsin Affiliated Hospitals Wauwatosa, WI
Sonya Dave, MD1, Lindsay Hammons, MD1, Alejandro Torres, MD1, Jeanne Hryciuk, MD2, Srivats Madhavan, MD1 1Medical College of Wisconsin Affiliated Hospitals, Wauwatosa, WI; 2Clement J. Zablocki Veterans Affairs Medical Center, Milwaukee, WI
Introduction: Langerhans Cell Histiocytosis (LCH) is a rare inflammatory myeloid disorder mostly seen in children before the age of five. The clinical presentation is heterogeneous with potential for wide-ranging organ involvement, primarily in the bone, skin, and lungs. Gastrointestinal tract involvement is very rare in LCH, and in symptomatic patients, etiology could be confused with infectious, allergic, and autoimmune bowel diseases. In children, LCH GI tract involvement has been associated with decreased survival due to multisystem involvement and digestive complications including diarrhea, vomiting, malabsorption, and failure to thrive. Review of literature shows less than twenty adults with incidences of GI tract involvement of LCH and only four cases of adults with LCH colon polyps.
Case Description/Methods: A 66-year-old man presented for screening colonoscopy. He reported generally good health without any GI or systemic symptoms or medical history. Family history was significant for leukemia in his brother. He currently smokes cigarettes and has a 2.5 pack-year history. On colonoscopy, six benign-appearing polyps were removed using cold snare polypectomy: three from the ascending colon (largest 0.5 cm in size), one from the splenic flexure of the transverse colon (0.4 cm in size), and two from the rectum (largest 0.4 cm in size). Quality of bowel preparation was excellent and the procedure was without complication. Rectal polyps were hyperplastic. Submucosal sections of the ascending and transverse colon polyps stained positive for CD1a, CD68, and S-100, consistent with Langerhans-type cells. The LCH diagnosis was further supported by irregular nuclei within focal nuclear grooves and an eosinophilic cytoplasm. Genomic testing of the colon sample revealed BRAFV600E mutation, the most recognized targetable mutation in LCH. He underwent further testing with PET scan and bone marrow biopsy, both of which were negative for LCH. Surveillance colonoscopy was recommended in 7 years.
Discussion: With his lack of systemic disease and asymptomatic presentation, watchful waiting and Oncology follow-up was recommended. Given the unusual diagnosis of LCH via a colon polyp, it is possible for lesions to be clinically unsuspected and histologically overlooked. With increasing rates of screening colonoscopy, more polyps demonstrating LCH on histopathology may be identified. Reports of LCH in the GI tract and colon polyps are infrequent and characterization of outcomes will rely on further cases in this population.
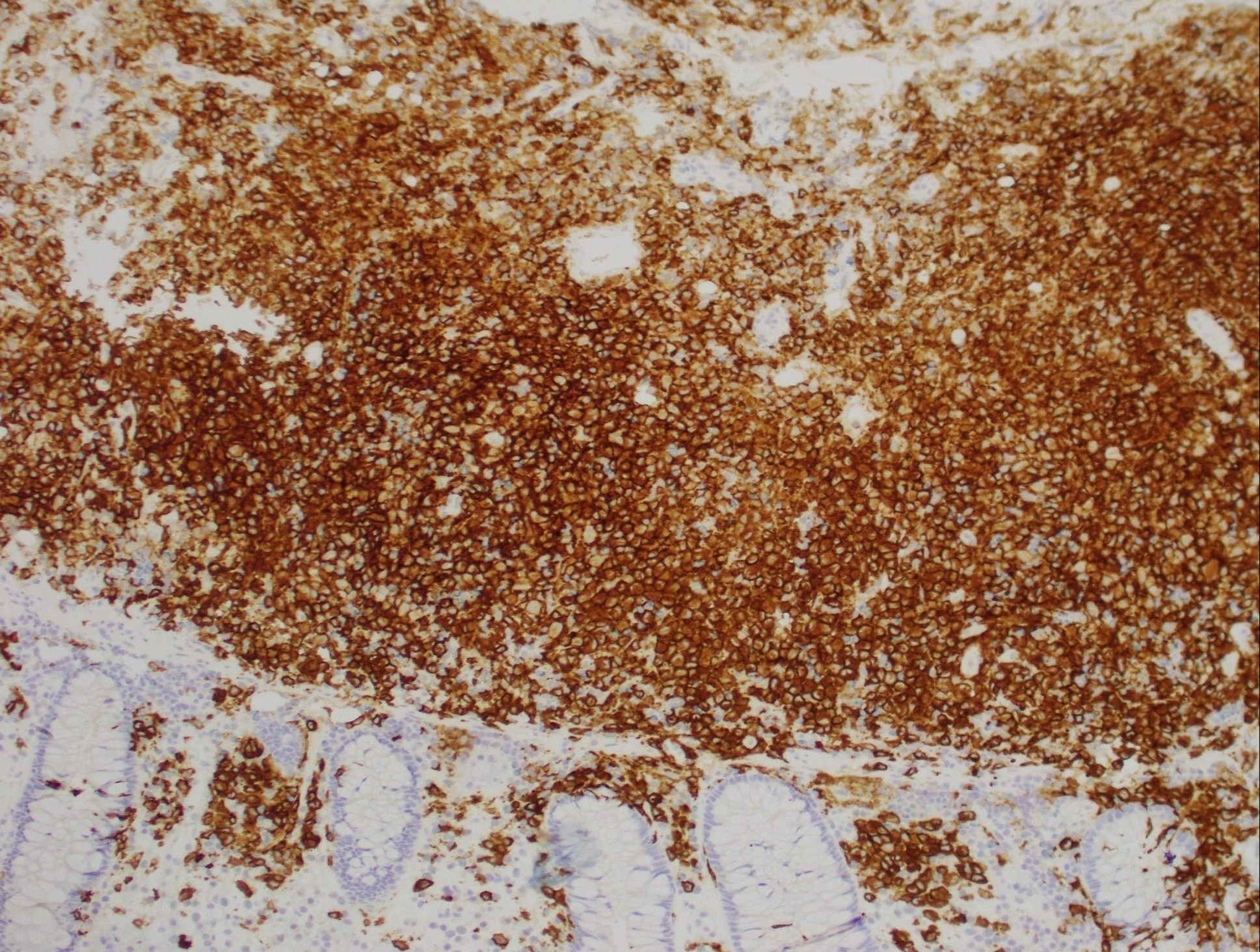
Figure: Positive immunostaining for CD1a, classically expressed by Langerhans cells.
Disclosures:
Sonya Dave indicated no relevant financial relationships.
Lindsay Hammons indicated no relevant financial relationships.
Alejandro Torres indicated no relevant financial relationships.
Jeanne Hryciuk indicated no relevant financial relationships.
Srivats Madhavan indicated no relevant financial relationships.
Sonya Dave, MD1, Lindsay Hammons, MD1, Alejandro Torres, MD1, Jeanne Hryciuk, MD2, Srivats Madhavan, MD1. C0158 - Rare Colonic Polyps: A Case of Langerhans Histiocytosis, ACG 2022 Annual Scientific Meeting Abstracts. Charlotte, NC: American College of Gastroenterology.