Louisiana State University Health Sciences Center Shreveport, LA
Ariana Bolumen, MD1, Kristie R. Searcy, MD1, Marlene Broussard, MD1, Christopher Oglesby, MD2, James Morris, MD, FACG3 1Louisiana State University Health Sciences Center, Shreveport, LA; 2LSUHSC, Shreveport, LA; 3LSU Health Sciences Center, Shreveport, LA
Introduction: Fulminant liver failure in the pediatric population remains an evolving topic due to its high impact on morbidity and mortality. Although, numerous causes have been proposed, 45% of cases remain cryptogenic. This abstract raise awareness of an incidence of fulminant hepatic failure secondary to Epstein Barr virus (EBV) infection in the presence of an unknown underlying rare immunological phenomenon, X-Linked Lymphoproliferative Syndrome (XLP).
Case Description/Methods: 17-year-old male with a past medical history of B Cell Lymphoma with colonic involvement at the age 3 presented with a 2-week history of fever, right upper quadrant abdominal pain, and jaundice associated with vomiting, diaphoresis, and weight loss. Pertinent positive labs included albumin 2.9 g/dL, T Bili 5.7 mg/dL, Alk Phos 806 U/L, AST 675U/L, ALT417 U/L, GGT 381 U/L, INR 1.58, Triglycerides 334 mg/dL, Ferritin 26,000 ng/mL, Fibrinogen 97 mg/dL. EBV PCR and flow cytometry confirmed an acute EBV infection. CT abdomen/pelvis showed hepatosplenomegaly, proctocolitis, and widespread lymphadenopathy. Liver U/S revealed gallbladder sludge without biliary dilatation.
The intractable fevers, hepatosplenomegaly and labs indicating coagulopathy, lactic acidemia, hypoglycemia, and hyperammonemia were concerning for acute liver failure requiring organ transplant. Pancytopenia, agammaglobulinemia, thrombocytopenia and hyperferritinemia suggested Hemophagocytic Lymphohistiocytosis (HLH) confirmed by bone marrow biopsy. Despite antibiotics, antifungals and immunosuppressive therapy—steroids, IVIG, Rituximab, Alemtuzumab administration, hepatic encephalopathy ensued leading to death. Post-mortem genetic panel was positive for UNC13D and SH2D1A gene mutation.
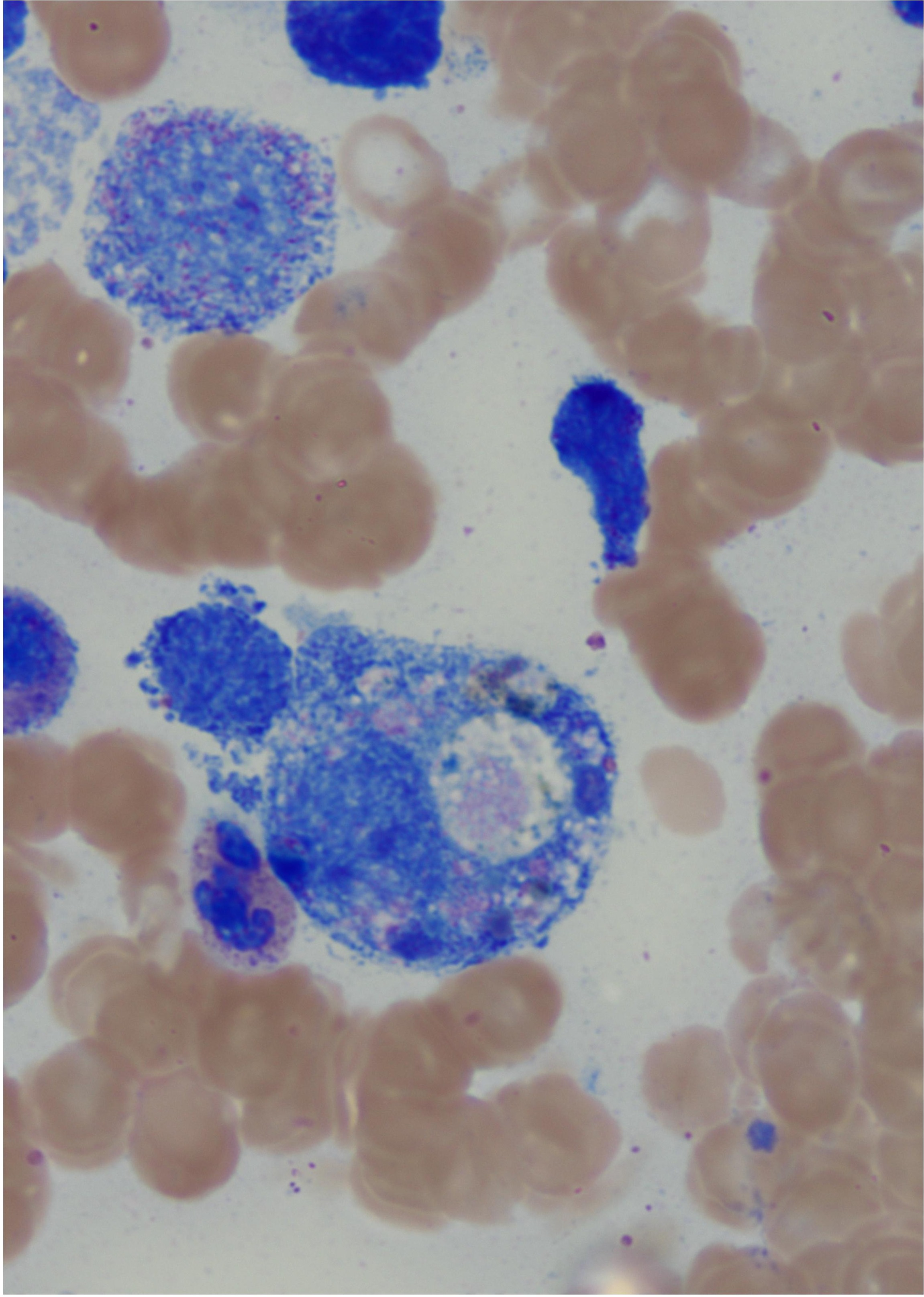
Discussion: EBV is commonly a self-limiting virus. In rare cases, EBV produces an alteration in NK and Cytotoxic T Cells which leads to the inability to properly eliminate macrophages and CD8+ T cells causing the immune dysregulation known as HLH. Suspicion for inherited HLH was supported by UNC13D gene mutation, hemophagocytes on bone marrow biopsy (Figure 1) and decreased levels of NK and T cells. This notion was challenged when genetic panel revealed a mutation in SH2D1A gene unique to XLP, which idiopathically causes sensitivity to EBV. XLP is linked with Non-Hodgkin’s B Cell Lymphomas manifesting extra-nodal in the intestinal tract. EBV-associated hepatitis and agammaglobulinemia w/ or w/o B cell malignancy should raise suspicion for XLP and prompt early initiation of treatment.
Figure: Figure 1 illustrates the numerous hemophagocytes seen on bone marrow biopsy supporting the diagnosis of Hemophagocytic Lymphohistiocytosis
Disclosures:
Ariana Bolumen indicated no relevant financial relationships.
Kristie Searcy indicated no relevant financial relationships.
Marlene Broussard indicated no relevant financial relationships.
Christopher Oglesby indicated no relevant financial relationships.
James Morris: Celgene – Grant/Research Support.
Ariana Bolumen, MD1, Kristie R. Searcy, MD1, Marlene Broussard, MD1, Christopher Oglesby, MD2, James Morris, MD, FACG3. A0615 - Fulminant Hepatitis Becomes the Initial Presentation of X-Linked Lymphoproliferative Disease, ACG 2022 Annual Scientific Meeting Abstracts. Charlotte, NC: American College of Gastroenterology.