Subhash Garikipati, MD1, Dhruvkumar Patel, MBBS2, Hrishikesh Samant, MD3 1Louisiana State University, Shreveport, LA; 2LSU Health Sciences Center, Shreveport, LA; 3Ochsner Medical Center, New Orleans, LA
Introduction: Cirrhosis in younger adults is often caused by autoimmune hepatitis, Wilson’s disease, and primary sclerosing cholangitis, but genetic-metabolic disorders must also be included in the differential. Lysosomal acid lipase deficiency (LAL-D) is a rare lysosomal storage disorder that can range from mild and presenting in early adulthood to severe with mortality within one year of life. The adult variant of LAL-D can sometimes be overlooked since symptoms are nonspecific, but it is important to recognize as enzyme replacement therapy can be life-saving.
Case Description/Methods: A 20-year-old male with no medical history presented with incidental transaminitis (AST 99U/L, ALT 99 U/L), thrombocytopenia (62,000/ul) and leukocytopenia (2200/ul) without any clinical symptoms. A bone marrow biopsy showed normal cellularity for the age group without evidence of clonal proliferation. Abdominal ultrasound revealed features suggestive of portal hypertension and fibroscan showed steatosis grade S3/fibrosis score F4. Next, a liver biopsy showed glycogenic hepatopathy with periportal and focal bridging fibrosis. Furthermore, an EGD showed possible villous blunting but biopsy ruled out celiac disease and there was no evidence of esophageal varices/portal gastropathy. Both pathologies did reveal clear foamy appearing cytoplasm that contain Period acid-Schiff-diastase stain positive. In addition, enzyme levels of liposomal acid lipase were 0.0 nmol/h/ml (normal > 21.0) and confirmatory LIPA gene sequencing was done and the patient was found to have liposomal acid lipase deficiency. He was also found to be heterozygous for alpha-1 antitrypsin (A1AT) deficiency of the M and Z isoforms with a level of 87 mg/dl. Treatment included sebelipase alfa 1 mg/kg infusions every 2 weeks.
Discussion: LAL-D is a rare, autosomal recessive lysosomal storage disease that involves a deficiency of LAL which is involved in the degradation of triglycerides and cholesterols, thereby causing accumulation of these substances in the cells. This deficiency is caused by a mutation in the LIPA gene. Our patient not only had the lysosomal storage disease causing liver injury but was possibly exacerbated by the addition of heterozygous A1AT deficiency which causes modest deficiency of the enzyme. These patients should be monitored for premature atherosclerotic vascular disease and hepatocellular carcinoma. Administering recombinant human LAL enzyme, sebelipase alfa, improves life expectancy.
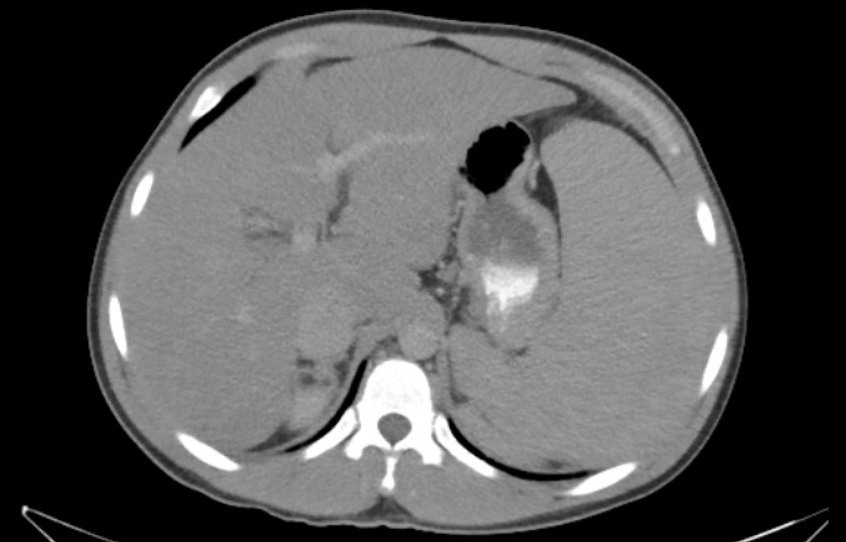
Figure: Diffuse hypodense liver suggestive of fatty liver disease and massive splenomegaly
Disclosures:
Subhash Garikipati indicated no relevant financial relationships.
Dhruvkumar Patel indicated no relevant financial relationships.
Hrishikesh Samant indicated no relevant financial relationships.
Subhash Garikipati, MD1, Dhruvkumar Patel, MBBS2, Hrishikesh Samant, MD3. D0528 - Two Concurrent Genetic Disorders Leading to Liver Cirrhosis, ACG 2022 Annual Scientific Meeting Abstracts. Charlotte, NC: American College of Gastroenterology.