Garvit Chhabra, MD1, Priyanshu Nain, MBBS2, Anuja Abhyankar, MD1, Navroop Nagra, MD1, Endashaw Omer, MD, MPH3 1University of Louisville, Louisville, KY; 2Medical College of Georgia, Augusta, GA; 3University of Louisville Hospital, Louisville, KY
Introduction: Primary hepatic angiosarcoma (PHA) is a rare aggressive endothelial cell tumor which is seen in patients in their 60s and 70s with 3:1 male predominance. Industrial exposure to vinyl chloride, radium, chronic arsenic ingestion, anabolic steroid use, and iatrogenic exposure to thorotrast radiocontrast are some of the known etiologies for the development of PHA. Whereas, in most cases, attributable risk factors are rarely identified.
Case Description/Methods: A 68-year-old male patient with NIDDM presented with chest pain and abdominal distension. Examination revealed marked ascites, abdominal tenderness, and bilateral leg edema. Labs were significant for thrombocytopenia and cholestatic pattern of liver enzymes with AST 94, ALT 47, ALP 413, T bilirubin 5.7. Paracentesis was negative for SBP or malignant cytology and was indicative of portal hypertensive ascites. Tumor marker workup was positive for CA 19-9 106.1, whereas negative for CEA 1.82, and AFP 2.7. CT A/P showed multifocal large heterogeneous enhancing lesions in the liver which were biopsied showing diffuse proliferation of abnormal vascular endothelial cells staining positive for CD34 and a diagnosis of PHA was made. With multifocal lesions, the patient was started on paclitaxel chemotherapy, which was eventually stopped with worsening liver function. The hospital course was complicated by hepatic encephalopathy and coagulopathy. The patient and the family later chose to transition to palliative therapy for comfort.
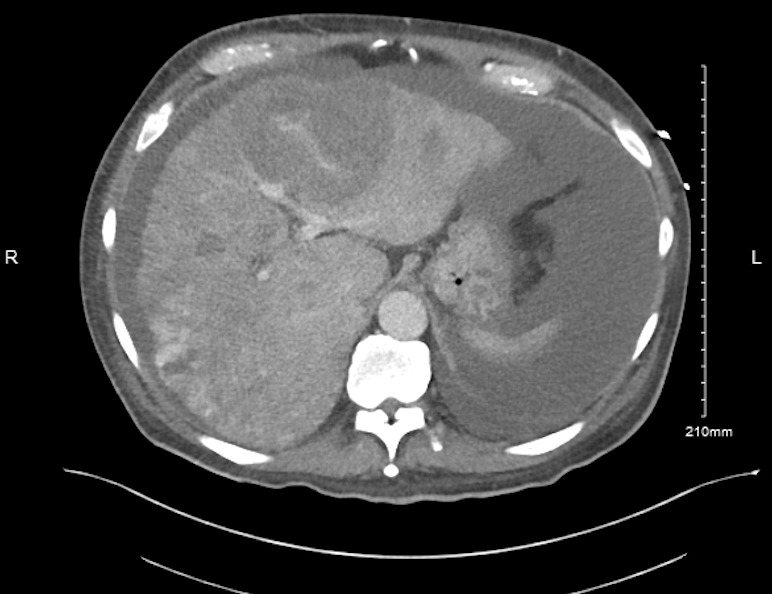
Discussion: PHA is a rare and aggressive tumor with poor outcomes and an average survival rate of less than a year. Early diagnosis is challenging as it presents with nonspecific abdominal symptoms. With progression, PHA can present as decompensated pseudocirrhosis due to compression of liver parenchyma leading to portal hypertension. Contrast-enhanced US and CT can help in diagnosis by showing lesions characteristic of central non-enhancement and peripheral irregular enhancement in the arterial and portal phase, and complete wash-out in the late phase. A definitive diagnosis of PHA is established via histopathological analysis with immunohistochemistry staining but can make diagnosis more challenging due to the potential of associated bleeding. Surgical excision with negative surgical margins is the standard treatment for PHA which is localized and resectable. Multifocal and metastatic PHA is a radio-resistant tumor with a paucity of treatment options, however, TACE and/or salvage chemotherapy can be potentially attempted.
Figure: CT Abdomen pelvis showing multiple mass lesions occupying most of liver parenchyma
Disclosures:
Garvit Chhabra indicated no relevant financial relationships.
Priyanshu Nain indicated no relevant financial relationships.
Anuja Abhyankar indicated no relevant financial relationships.
Navroop Nagra indicated no relevant financial relationships.
Endashaw Omer indicated no relevant financial relationships.
Garvit Chhabra, MD1, Priyanshu Nain, MBBS2, Anuja Abhyankar, MD1, Navroop Nagra, MD1, Endashaw Omer, MD, MPH3. E0551 - Primary Hepatic Angiosarcoma: A Rare Cause of Decompensated Pseudo-Cirrhosis and Acute Liver Failure, ACG 2022 Annual Scientific Meeting Abstracts. Charlotte, NC: American College of Gastroenterology.